Лечение т-клеточный лейкоз из больших гранулярных лимфоцитов
ФГБУ «Гематологический научный центр» Минздрава России, Москва
ФГБУ «Гематологический научный центр» Минздрава России, Москва
ГБОУ ВПО «Первый МГМУ им. И.М. Сеченова» Минздрава России, Москва, Россия
Кафедра хирургии ФГБОУ ДПО «Российская медицинская академия непрерывного профессионального образования» Минздрава РФ, Москва, Россия
ФГБУ Гематологический научный центр Минздрава России, Москва, Россия
ФГБУ Гематологический научный центр Минздрава России, Москва, Россия
ФГБУ «Гематологический научный цент» Минздрава России, Москва
ФГБУ «Гематологический научный центр» Минздрава России, Москва
ГБОУ ДПО РМАПО Минздрава России, Москва, Россия
ФГБУ «Гематологический научный центр» Минздрава России, Москва
ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России, 125167, Москва, Россия
Гематологический научный центр Минздрава России, Москва
Научно-клиническое отделение гематологической хирургии Гематологического научного центра Минздрава РФ, Москва
Гематологический научный центр Минздрава России
ГБОУ ВПО «Первый МГМУ им. И.М. Сеченова» Минздрава России, Москва
Гематологический научный центр Минздрава России, Москва
ГНЦ Минздрава России, Москва
ФГБУ Гематологический научный центр Минздрава России, Москва, Россия
Гематологический научный центр Минздрава РФ, Москва, Россия
Гематологический научный центр Минздрава России, Москва
ФГБУ «Гематологический научный центр» Минздрава России, Москва, Россия
ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России, Москва, Россия ,
Гепатолиенальная Т-клеточная лимфома: проблемы диагностики и лечения
Чернова Н.Г., Джулакян У.Л., Виноградова Ю.Е., Сидорова Ю.В., Рыжикова Н.В., Коржова С.М., Синицына М.Н., Тихомиров Д.С., Наумова Е.В., Обухова Т.Н., Двирнык В.Н., Судариков А.Б., Ковригина А.М., Кравченко С.К., Меликян А.Л., Кузьмина Л.А., Гальцева И.В., Смирнова С.Ю., Гемджян Э.Г., Звонков Е.Е., Паровичникова Е.Н., Савченко В.Г. Гепатолиенальная Т-клеточная лимфома: проблемы диагностики и лечения. Терапевтический архив. 2016;88(7):4‑14.
Chernova NG, Dzhulakian UL, Vinogradova YuE, Sidorova YuV, Ryzhikova NV, Korzhova SM, Sinitsyna MN, Tikhomirov DS, Naumova EV, Obukhova TN, Dvirnyk VN, Sudarikov AB, Kovrigina AM, Kravchenko SK, Melikjan AL, Kuz’mina LA, Gal’tseva IV, Smirnova SYu, Gemdzhyan EG, Zvonkov EE, Parovichnikova EN, Savchenko VG. Hepatosplenic T-cell lymphoma: The problems of diagnosis and treatment. Terapevticheskii Arkhiv. 2016;88(7):4‑14. (In Russ.)
https://doi.org/10.17116/terarkh20168874-14
Аннотация За последнее десятилетие достигнуты значительные успехи в понимании патогенеза NK/Т-клеточных лимфом, однако их диагностика остается затруднительной вследствие их редкости и клинико-морфологического разнообразия. В статье обобщен десятилетний опыт диагностики и лечения гепатолиенальной Т-клеточной лимфомы (ГЛТЛ) в ФГБУ «Гематологический научный центр» Минздрава России, рассмотрены вопросы дифференциальной диагностики с другими гематологическими заболеваниями, протекающими со сходными клинико-лабораторными симптомами, сформулированы современные подходы к диагностике и лечению. Представлен взгляд клинициста на проблему диагностики и лечения данного заболевания. Показано, что ГЛТЛ — гетерогенная группа заболеваний, различающихся по реаранжировкам генов цепей Т-клеточного рецептора, клиническому течению заболевания, общей выживаемости (ОВ). По нашим данным, 3-летняя ОВ составила 12%, медиана продолжительности жизни — 26 мес. Для вариантов γδ и αβ ГЛТЛ 2-летняя ОВ равна 25 и 70% соответственно. Различие ОВ для вариантов ГЛТЛ не достигало статистической значимости (возможно, в силу недостаточного объема выборки).
ФГБУ «Гематологический научный центр» Минздрава России, Москва
ФГБУ «Гематологический научный центр» Минздрава России, Москва
ГБОУ ВПО «Первый МГМУ им. И.М. Сеченова» Минздрава России, Москва, Россия
Кафедра хирургии ФГБОУ ДПО «Российская медицинская академия непрерывного профессионального образования» Минздрава РФ, Москва, Россия
ФГБУ Гематологический научный центр Минздрава России, Москва, Россия
ФГБУ Гематологический научный центр Минздрава России, Москва, Россия
ФГБУ «Гематологический научный цент» Минздрава России, Москва
ФГБУ «Гематологический научный центр» Минздрава России, Москва
ГБОУ ДПО РМАПО Минздрава России, Москва, Россия
ФГБУ «Гематологический научный центр» Минздрава России, Москва
ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России, 125167, Москва, Россия
Гематологический научный центр Минздрава России, Москва
Научно-клиническое отделение гематологической хирургии Гематологического научного центра Минздрава РФ, Москва
Гематологический научный центр Минздрава России
ГБОУ ВПО «Первый МГМУ им. И.М. Сеченова» Минздрава России, Москва
Гематологический научный центр Минздрава России, Москва
ГНЦ Минздрава России, Москва
ФГБУ Гематологический научный центр Минздрава России, Москва, Россия
Гематологический научный центр Минздрава РФ, Москва, Россия
Гематологический научный центр Минздрава России, Москва
ФГБУ «Гематологический научный центр» Минздрава России, Москва, Россия
ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России, Москва, Россия ,
алло-ТГСК — трансплантация аллогенных гемопоэтических стволовых клеток
алло-ТКМ — трансплантация аллогенного костного мозга
БГЛ — хронический Т-клеточный лейкоз из больших гранулярных лимфоцитов
ВКЛ — волосатоклеточный лейкоз
ГЛТЛ — гепатолиенальная Т-клеточная лимфома
ГНЦ — ФГБУ Гематологический научный центр Минздрава России
ЖКБ — желчнокаменная болезнь
ЗР — зрелые реаранжировки
ИГХИ — иммуногистохимическое исследования
ИФИ — иммунофенотипическое исследование
КМ — костный мозг
ЛУ — лимфатические узлы
МДС — миелодиспластический синдром
ОВ — общая выживаемость
ПЦР — полимеразная цепная реакция
РР — ранние реаранжировки
ЩФ — щелочная фосфатаза
TCR — Т-клеточный рецептор
TCRB — цепь β Т-клеточного рецептора
TCRD — цепь δ Т-клеточного рецептора
TCRG — цепь γ Т-клеточного рецептора
NK/Т-клеточные лимфомы — группа лимфопролиферативных заболеваний, составляющая 10—15% всех неходжкинских лимфом [1]. За последнее десятилетие достигнуты значительные успехи в понимании патогенеза Т-клеточных лимфом, однако их диагностика представляет трудности. Прежде всего это связано с их редкостью и клинико-морфологическим разнообразием [2, 3]. Гепатолиенальная Т-клеточная лимфома (ГЛТЛ) — лимфопролиферативное заболевание, характеризующееся преимущественным поражением печени, селезенки и костного мозга (КМ) [4]. Первое описание ГЛТЛ опубликовано в 1990 г. J. Farcet и соавт. [5], где представлены клинико-морфологические особенности лимфопролиферативного процесса, протекающего с поражением печени, селезенки и К.М. Заболевание дебютировало с гепатоспленомегалии, тромбоцитопении. При морфологическом исследовании выявлены инфильтрация синусоид печени, селезенки и КМ Т-клетками CD3 + /TCRγδ. Несмотря на лечение, наблюдалось быстропрогрессирующее течение лимфомы.
Термин «γδ-гепатолиенальная Т-клеточная лимфома» включен в качестве предварительной нозологической единицы в REAL-классификацию 1994 г. [6]. В последствии выявлен вариант αβ-заболевания, и в классификации ВОЗ от 2008 г. заболевание обозначено термином «гепатолиенальная Т-клеточная лимфома» [4].
Гепатолиенальная лимфома составляет лишь 1,4% среди зрелых NK/Т-клеточных лимфом [2]. Чаще заболевают молодые мужчины в возрасте около 40 лет [7]. Приблизительно в 10—20% случаев ГЛТЛ отмечается у больных с вторичным иммунодецифитным состоянием, связанным с трансплантацией солидных органов или гемопоэтических клеток, лечением других предшествовавших неоплазий, иммуносупрессивной терапией аутоиммунных заболеваний, перенесенной малярией [8—11].
Клиническая картина ГЛТЛ характеризуется гепатоспленомегалией, нарушением функции печени, анемией, тромбоцитопенией [12]. Преобладают жалобы на сильную слабость, лихорадку неправильного типа, ночную потливость, снижение массы тела, тяжесть или боли в левом и правом подреберьях. В гемограмме выявляются лейкопения, анемия, тромбоцитопения. В половине случаев обнаруживается повышение активности лактатдегидрогеназы (ЛДГ). При выраженном поражении печени может наблюдаться нарушение белково-синтетической функции. Развитие ГЛТЛ обычно не ассоциировано с реактивацией вирусной инфекции [7]. Вовлечение К.М. наблюдается у 2/3 пациентов, а лейкемизация опухолевого процесса обнаруживается в 1/3 случаев [13].
Диагноз ГЛТЛ устанавливают на основании исследования биоптата опухоли. Морфологическое исследование КМ выявляет опухолевую интерстициальную и/или интрасинусоидальную инфильтрацию атипичными лимфоидными клетками мелких, средних и крупных размеров с ядрами неправильной формы. В гистологических препаратах, окрашенных гематоксилином и эозином, опухолевые клетки не всегда хорошо различимы среди клеток миелоидной ткани, их позволяет выявить иммуногистохимическое окрашивание. Опухолевые клетки экспрессируют CD2, CD3, CD56, некоторые цитотоксические молекулы TIA-1, гранзим М, обычно не экспрессируют ни CD4, ни CD8, а также отсутствуют CD5, CD7, перфорин и гранзим В. Экспрессия NK-клеточных маркеров CD16 и CD56 на поверхности опухолевых клеток вариабельна. Морфологическое исследование селезенки и печени выявляет инфильтрацию опухолевыми клетками красной пульпы селезенки и синусоид печени [14—15].
В зависимости от типа Т-клеточного рецептора (TCR) выделяют два основных варианта ГЛТЛ: γδ и αβ. В случаях варианта γδ ГЛТЛ наряду с перестройками генов цепей δ и γ TCR (TCRD, TCRG) может обнаруживаться незавершенная или непродуктивная реаранжировка генов цепи β TCR (TCRB) [16, 17]. Несмотря на то что варианты γδ и αβ ГЛТЛ объединены в рамках одной нозологической единицы, они демонстрируют клинико-морфологические различия. Так, у молодых мужчин и пациентов с иммуносупрессией в анамнезе в 80% случаев диагностируется вариант γδ-заболевания. Опухолевые клетки при варианте γδ ГЛТЛ в большинстве случаев не экспрессируют антиген CD8, тогда как при варианте αβ он выявляется.
При цитогенетическом исследовании чаще наблюдаются аномалии 7-й хромосомы: изохромосома 7q или кольцевая 7-я хромосома, реже встречается трисомия 8-й хромосомы или потеря половых хромосом [18—20].
Клиническое течение ГЛТЛ в большинстве случаев агрессивное. Известно, что вариант γ ГЛТЛ характеризуется более агрессивным течением и крайне неблагоприятным прогнозом подобно другим Т-клеточным лимфомам γδ. Большинство пациентов умирают в течение 1 года, а 5-летняя общая выживаемость (ОВ) составляет лишь 7% [2].
Оптимальная тактика лечения больных с этим видом Т-клеточной лимфомы не разработана. Представленный в источниках литературы опыт по лечению ГЛТЛ крайне разнообразен, что обусловлено поиском наиболее эффективной комбинации противоопухолевых препаратов. Лечение с применением схем, содержащих антрациклины, малоэффективно, отмечаются кратковременность полных ремиссий, высокая частота ранних рецидивов. Кроме курсов, подобных СНОР, применялись платиносодержащие курсы, высокие дозы цитарабина, аналоги пурина (пентостатин), Hyper-CVAD/hdAra-C и hdМТХ [13, 21, 22]. Наилучшие результаты достигнуты при проведении консолидирующей трансплантации аллогенных гемопоэтических кроветворных клеток (алло-ТГСК) в первой полной ремиссии [7].
К факторам, ассоциированным с неблагоприятным прогнозом, относят мужской пол, отсутствие полной ремиссии заболевания после индукционной химиотерапии (ХТ), наличие иммуносупрессивной терапии в анамнезе, а также отсутствие реаранжировок генов цепи γ TCR [7, 8].
Материалы и методы
Пациенты. Проведен ретроспективный анализ 18 пациентов (6 мужчин, 12 женщин) в возрасте от 24 до 76 лет, медиана 52 года с диагностированной ГЛТЛ в ФГБУ ГНЦ Минздрава России с 2005 по 2015 г. Клиническая характеристика пациентов представлена в табл. 1.

Таблица 1. Клинико-лабораторная характеристика обследованных больных
Проанализированы клинические признаки, данные морфологического, иммунофенотипического и молекулярного исследований, а также эффективность индукционной терапии и общая выживаемость.
Методы диагностики. Всем больным проведено полное клинико-лабораторное обследование. Для патоморфологической верификации ГЛТЛ выполняли гистологическое и иммуногистохимическое исследования (ИГХИ) с расширенной панелью моноклональных антител (CD2, CD3, CD4, CD5, CD7, CD8, CD16, CD56, TCR (γ), TCR (bF1), Granzyme B, TIA-1, Ki-67).
Иммунофенотипическое исследование (ИФИ) образцов КМ, периферической крови и селезенки методом многоцветной проточной цитометрии проводили на восьмицветном проточном цитометре FACS Canto II («Becton Dickinson», США) или десятицветном проточном цитометре Navios («Beckman Coulter», США) c использованием реагентов производства фирм «Becton Dickinson», «BeckmanCoulter» и «Dako». Диагностические панели включали CD2, CD3, CD4, CD5, CD7, CD8, CD16, CD19, CD45, CD56, CD57, TCRαb, TCRgd.
Оценку T-клеточной клональности проводили по реаранжировкам генов TCRD (Vδ-Dδ, Dδ-Dδ, Dδ-Jδ, Vδ-Jδ), TCRG (Vγ-Jγ) и TCRB (Vβ-Jβ, Dβ-Jβ). Для этого использовали метод полимеразной цепной реакции (ПЦР) с мультиплексными системами праймеров BIOMED-2 [23] и последующий фрагментный анализ на секвенаторе ABI PRISM 3130 Genetic Analyzer («Applied Biosystems», CША). Флуоресценция амплификатов и их профиль (распределение по длинам) оценивали при помощи компьютерной программы GeneMapper v. 4.0 («Applied Biosystems», США).
Содержание иммуноглобулинов класса G, А, М, а также С-реактивного белка и β 2 -микроглобулина в сыворотке крови определяли методом кинетической нефелометрии (нефелометр Immage 800, «Beckman Coulter», США).
При стандартном цитогенетическом исследовании клетки КМ, периферической крови и селезенки культивировали в питательной среде RPMI 1640 с добавлением 20% эмбриональной телячьей сыворотки, глютамина и антибиотиков 24 ч без и 72 ч с добавлением фитогемагглютинина. G-дифференциальное окрашивание хромосом осуществляли с использованием краски Wright («Merck»). При возможности анализировали 20 метафаз. Кариотип описывали в соответствии с Международной номенклатурой хромосом человека ISCN, 2013 [24].
Эффективность противоопухолевого ответа определяли согласно международным критериям [25].
С лечебно-диагностической целью больным выполняли спленэктомию (СЭ) [26] и биопсию печени.
Анализ ОВ проведен по методу Каплана—Мейера. При расчете ОВ время отсчитывали от установки диагноза до летального исхода (или последней информации о больном). Различия при р
Результаты
В нашем исследовании у 11 (61%) из 18 больных первоначально установлены ошибочные диагнозы: миелодиспластический синдром (МДС) — у 3, волосатоклеточный лейкоз (ВКЛ) — у 3, хронический Т-клеточный лейкоз из больших гранулярных лимфоцитов (БГЛ) — у 3, периферическая Т-клеточная лимфома, неспецифицированная — у 1. Большинство пациентов поступали в клиники ГНЦ в крайне тяжелом состоянии, обусловленном распространенным опухолевым процессом с выраженными В-симптомами, иммунными осложнениями и инфекционными заболеваниями.
В табл. 1 представлена клинико-лабораторная характеристика обследованных больных. В большинстве случаев они предъявляли жалобы на постоянные боли в левом подреберье, а в 14 случаях при ультразвуковом исследовании выявлены инфаркты селезенки.
У всех пациентов выявлена гепатоспленомегалия, размеры селезенки варьировали от 14 см до гигантских (25 см и более). Вовлечения периферических лимфатических узлов (ЛУ) не отмечено, а в 8 (44%) случаях выявлялось незначительное увеличение размеров регионарных ЛУ в воротах печени и селезенки.
При анализе сопутствующей патологии выявлено 2 случая ревматоидного артрита, для лечения которого длительно применялась иммуносупрессивная терапия метотрексатом, у 1 больного хронический вирусный гепатит С, у 6 желчнокаменная болезнь (ЖКБ). Ни один пациент не являлся реципиентом солидных органов или гемопоэтических клеток.
Изменения гемограммы отмечались у всех больных: анемия (гемоглобин менее 120 г/л) у 15 (83%), лейкопения (количество лейкоцитов менее 4·10 9 /л) у 14 (78%), тромбоцитопения (количество тромбоцитов менее 100·10 9 /л) у 13 (72%). В биохимическом анализе крови повышение активности ЛДГ обнаружено у 65% больных, щелочной фосфатазы (ЩФ) у 100%. Признаки внутрисосудистого гемолиза (гипербилирубинемия за счет непрямой фракции, ретикулоцитоз, выявление антиэритроцитарных антител) наблюдались у 14 из 18 пациентов. Гипербилирубинемия за счет непрямой фракции выявлена в 10 (59%) из 18 случаев, ретикулоцитоз в 9 (90%) из 10, прямая проба Кумбса выполнена в 8 случаях, в 2 получен положительный результат.
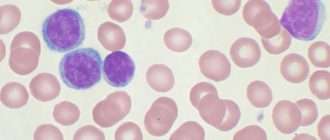
Во всех случаях выполнено цитологическое исследование пунктата КМ и мазков периферической крови. В половине случаев опухолевые лимфоциты были крупнее обычных, около 16—18 мкм, с округлым или плеоморфным ядром, омоложенной структурой хроматина, содержащим одну или несколько нуклеол, с неровным краем базофильной цитоплазмы (рис. 1). Ни в одном случае не обнаружено лимфоцитов с крупными азурофильными гранулами, характерными для хронического Т-клеточного лейкоза из больших гранулярных лимфоцитов, в ряде случаев в лимфоидных клетках выявляли мелкую азурофильную зернистость.

Рис. 1. Мазок периферической крови, окраска по Романовскому—Гимзе, ув. 100. Стрелками указаны опухолевые лимфоциты.
Гистологическое исследование трепанобиоптата КМ проведено всем больным, специфическое поражение КМ выявлено только в 14 (78%) случаях (рис. 2, а, б).

Рис. 2. Гистологическая картина селезенки, биоптатов печени и К.М. Окраска эозином и гематоксилином. а — биоптат КМ, ув. 100; б — биоптат КМ, ув. 400; в — селезенка, ув. 400; г — биоптат печени, ув. 400. Стрелками указано внутрисинусоидальное скопление опухолевых лимфоцитов.
СЭ и биопсию печени выполнили у 17 из 18 пациентов. Гистологическое исследование биоптатов селезенки позволило выявить стертость архитектоники строения за счет лимфоидной инфильтрации красной пульпы, множественные крупноочаговые разрастания опухолевых клеток средних и крупных размеров с ядрами округло-овальной или неправильной формы. При гистологическом исследовании биоптатов печени в синусоидных капиллярах выявлялась лимфоидноклеточная инфильтрация (рис. 2, в, г).
ИГХИ с применением расширенной панели моноклональных антител позволило верифицировать иммунофенотип и вариант TCR опухолевых клеток. Данные ИГХИ представлены в табл. 2.

Таблица 2. Результаты ИГХИ селезенки Примечание. *— СЭ не выполнялась, представлены данные ИГХИ КМ; ** — вариант αβ верифицирован по данным проточной цитометрии; здесь и в табл. 3—5: н/д — нет данных.
Во всех случаях наблюдалась экспрессия маркера CD3ε, экспрессия других кластеров дифференцировки варьировала. У 3 больных отмечена экспрессия CD4, в 6 случаях опухолевые клетки демонстрировали CD8-позитивный иммунофенотип, в остальных 9 случаях экспрессия маркеров CD4, CD8 не наблюдалась. Определение варианта ГЛТЛ проведено в 12 случаях, у 8 (67%) больных выявлен вариант αβ ГЛТЛ, у 4 (33%) — γδ. Экспрессия Ki67 варьировала от 5 до 30%.
ИФИ клеток КМ, периферической крови, селезенки методом проточной цитометрии выполнено у 14 из 18 пациентов. Основными показаниями к проведению проточной цитометрии служили верификация распространенности процесса и иммунофенотипическая характеристика опухолевых клеток. При исследовании экспрессии маркеров CD2, CD5, CD7 наблюдалась ее гетерогенность. В 2 случаях наряду с популяцией CD4 – /CD8 + опухолевых клеток выявлен клон с коэкспрессией CD4/CD8, составляющий 17 и 25% от общего числа Т-лимфоцитов. Иммунофенотипирование лимфоцитов периферической крови проведено у 5 пациентов, в 4 случаях верифицирована лейкемизация опухолевого процесса.
Определение T-клеточной клональности методом ПЦР проведено у 16 из 18 пациентов. Моноклональность Т-лимфоцитов по реаранжировкам генов цепей TCR выявлена во всех 16 исследованных случаях (табл. 3).

Таблица 3. Исследование Т-клеточной клональности по реаранжировкам генов TCRD, TCRG и TCRB Примечание. км — костный мозг; моно — выявлена моноклональная картина; поли — моноклон не выявлен, поликлональная картина; без с/э — без СЭ; РР — ранние реаранжировки; ЗР — зрелые реаранжировки. * — пациенты с ранним спектром клональных реаранжировок, характерным для Т-клеточных лимфом γδ (обнаружена клональная реаранжировка генов TCRD и/или отсутствует полная (Vβ—Jβ) клональная реаранжировка генов TCRB); ** — вариант TCR установлен по данным ИГХИ/ИФИ.
Молекулярное исследование реаранжировок генов TCRG проведено в 14 образцах периферической крови больных ГЛТЛ, в 10 (71%) случаях выявлена моноклональная картина, свидетельствующая о персистенции опухолевого клона в периферической крови, т. е. лейкемизации. Клональные реаранжировки генов TCRD выявлены у 7 из 16 обследованных пациентов (у 6 в селезенке и у 1 в КМ). У 3 пациентов обнаружены отличия клональных продуктов генов TCRB в селезенке и КМ, что демонстрирует наличие нескольких опухолевых клонов у данных пациентов. В 3 из 4 случаев выявлены молекулярные маркеры вовлечения КМ в отсутствие морфологических признаков.
Иммунохимическое исследование белков сыворотки крови у 11 (61%) больных выявило увеличение количества иммуноглобулинов хотя бы одного класса: IgG (норма 95—235 МЕ/мл) в 6 (33%), IgА (норма 55—250 МЕ/мл) в 9 (50%), IgM (норма 55—250 МЕ/мл) в 2 (11%) случаях. Данные иммунохимического анализа представлены в табл. 1. Увеличение количества циркулирующих иммунных комплексов отмечено у 6 (60%) из 10 пациентов, а у 7 из 8 пациентов — увеличение концентрации β 2 -микроглобулина, у 7 (70%) из 10 пациентов — С-реактивного белка.
Стандартное цитогенетическое исследование клеток КМ, периферической крови и селезенки выполнено у 13 из 18 больных, данные представлены в табл. 4.

Таблица 4. Стандартное цитогенетическое исследование клеток периферической крови, КМ, селезенки
Нарушений кариотипа не выявлено в 9 случаях, в одном случае не обнаружено делящихся клеток. В 3 случаях выявлены клональные хромосомные аберрации: у одного больного в клетках КМ обнаружена трисомия 21-й хромосомы; у второго больного в клетках крови выявлена t (6;15)(q21;q24—25), а в клетках селезенки та же транслокация в основном клоне и трисомия 8-й хромосомы в субклоне; в третьем случае в клетках селезенки наблюдались два клона с дериватом 19-й хромосомы и дополнительной маркерной хромосомой, в одной метафазе обнаружена другая дополнительная маркерная хромосома.
Лечение. Больным в исследуемой группе проведено лечение по различным протоколам, данные представлены в табл. 5.

Таблица 5. Лечение Примечание. СНОР (циклофосфамид, винкристин, преднизолон, адриамицин), А-СНОР (СНОР с алемтузумабом), СОР (циклофосфамид, винкристин, преднизолон), ld МТХ (метотрексат 50 мг еженедельно), ld МП (меркаптопурин 50 мг ежедневно), ldЦф (циклофосфан 50 мг ежедневно), HyperCVAD (циклофосфамид, винкристин, адриамицин, дексаметазон), hdAra-C (высокие дозы цитарабина), hd MTX (высокие дозы метотрексата), ОЛЛ, 2009, GMALL — программы терапии острых лимфобластных лейкозов. ЧР — частичная ремиссия; ПР — полная ремиссия; * — проведена трансплантация аллогенных гемопоэтических клеток.
У 13 из 18 пациентов до начала ХТ выполнена С.Э. Операцию проводили с целью уменьшения опухолевой массы, купирования иммунных нарушений, коррекции цитопении и уточнения диагноза. Масса удаленных селезенок составила в среднем 1950 (700—4500) г. В послеоперационном периоде у всех больных отмечалось восстановление показателей периферической крови. В некоторых случаях наблюдался аспленический тромбоцитоз до 1500·10 9 /л. В 4 случаях СЭ проведена с диагностической целью после неэффективного цитостатического лечения по поводу ошибочно установленных других видов гемобластозов. Следует отметить, что один из этих 4 пациентов получал лечение от предполагаемого волосатоклеточного лейкоза интерфероном-α (ИФН-α) и кладрибином в течение 10 лет до верификации диагноза варианта αβ ГЛТЛ.
В одном случае СЭ не проводилась, диагноз варианта αβ ГЛТЛ верифицирован по биоптату К.М. Принимая во внимание тяжесть состояния больной, обусловленную выраженными В-симптомами, гепатоспленомегалией, анемией до 70 г/л, лейкопенией менее 1·10 9 /л, пожилой возраст — 76 лет, высокий риск развития пневмонии в раннем послеоперационном периоде, операцию отложили и назначили лечение ИФН-α в дозе 3 млн ЕД 3 раза в неделю. В результате лечения состояние больной постепенно улучшалось. При обследовании через 12 мес постоянной терапии ИФН-α отмечены нормализация размеров селезенки, регрессия лимфоидной инфильтрации в биоптате КМ, восстановление количества лейкоцитов, гемоглобина. Методом ПЦР с последующим проведением капиллярного электрофореза, Т-клеточной клональности по реаранжировкам генов TCRG в образцах крови и КМ не выявлено. Несмотря на достижение клинико-гематологической ремиссии, пациентка продолжает лечение ИФН-α, срок наблюдения на момент написания статьи составляет 15 мес.
Шести пациентам с вариантами αβ и γδ ГЛТЛ проведена короткоимпульсная ХТ: СНОР (циклофосфамид, винкристин, преднизолон, адриамицин), А-СНОР (СНОР с алемтузумабом), СОР (циклофосфамид, винкристин, преднизолон), HyperCVAD (циклофосфамид, винкристин, адриамицин, дексаметазон)/hdAra-C (высокие дозы цитарабина) и hdMTX (высокие дозы метотрексата). Однако ни в одном случае не получено клинико-гематологической ремиссии заболевания. Трем пациентам 44, 55, 65 лет назначена терапия по программам ОЛЛ, 2009 [27] и GMALL [28]. Эти химиотерапевтические режимы, успешно применяемые для лечения острых лимфобластных лейкозов, предполагают длительное неинтенсивное цитостатическое воздействие. У пациентки 44 лет с вариантом αβ ГЛТЛ в результате терапии по программе GMALL получена клинико-гематологическая ремиссия заболевания. С консолидирующей целью проведена высокодозная ХТ по программе ВЕАМ с трансплантацией аутологичных гемопоэтических клеток. Однако через 2 мес появилась и стала прогрессировать цитопения, при молекулярном исследовании выявлены наблюдаемые в дебюте заболевания клональные реаранжировки генов TCRG и TCRB. Пациентке проведена трансплантация аллогенного КМ (алло-ТКМ) от родственного донора, в результате чего достигнута полная ремиссия заболевания. На момент написания статьи продолжительность наблюдения после алло-ТКМ составляет 12 мес, признаков рецидива заболевания нет. Пациент 55 лет с не уточненным вариантом ГЛТЛ, получавший терапию по программе ОЛЛ, 2009, умер от инфекционных осложнений вне прогрессирования лимфомы. У пациента 65 лет с вариантом γδ ГЛТЛ получена полная ремиссия в результате проведения 6 поддерживающих курсов по протоколу ОЛЛ, 2009. После завершения лечения в биоптате КМ наблюдалась регрессия опухолевой инфильтрации КМ, при молекулярном исследовании образцов КМ не выявлялись наблюдаемые ранее клональные реаранжировки генов TCRG. Продолжительность ремиссии составила 8 мес, впоследствии развился генерализованный рецидив с гепатомегалией, тромбоцитопенией и лейкемизацией опухолевого процесса. Учитывая возраст пациента, наличие сопутствующего заболевания сердца, сахарного диабета, назначили длительную терапию малыми дозами цитостатических препаратов, позволившую добиться стабилизации заболевания и продлить жизнь на 13 мес.
Длительная терапия малыми дозами цитостатических препаратов предполагает применение меркаптопурина 50 мг ежедневно, циклофосфамида 50 мг ежедневно, метотрексата 50 мг еженедельно в качестве моно- или комбинированной терапии. Длительная терапия малыми дозами цитостатических препаратов применена у 7 больных с вариантами αβ и с γδ ГЛТЛ и эффективна как в качестве индукционного лечения, так и при прогрессировании ГЛТЛ после короткоимпульсной терапии. Например, длительная терапия циклофосфамидом 50 мг ежедневно успешно проводилась пациентке 51 года с вариантом γδ ГЛТЛ и позволила достигнуть частичной ремиссии продолжительностью 22 мес. Однако полной эрадикации опухолевого клона на фоне длительной терапии малыми дозами цитостатических препаратов не достигнуто ни в одном случае. Необходимо отметить меньшее число инфекционных и токсических осложнений длительной терапии малыми дозами цитостатических препаратов, чем короткоимпульсной интенсивной терапии.
При анализе ОВ учтены данные только 17 больных, не учтены данные 1 больного в связи с отсутствием дальнейшего контакта. Трехлетняя О.В. для исследуемой группы больных ГЛТЛ составила 12% (10%), медиана продолжительности жизни — 26 мес (медиана периода наблюдения 18 мес), для вариантов γδ и αβ ГЛТЛ двухлетняя ОВ составила 25% (17%) и 70% (15%) соответственно. Различие О.В. вариантов ГЛТЛ не достигало статистической значимости, возможно, в силу недостаточного размера выборки (рис. 3).

Рис. 3. Трехлетняя О.В. 17 больных ГЛТЛ в целом (а) и двухлетняя OВ в зависимости от варианта TCR: αβ (n=8) и γδ (n=4) (б).
Таким образом, короткоимпульсная ХТ по программам СНОР, А-СНОР, СОР, HyperCVAD/hdAra-C и hdMTX показывает низкую эффективность при лечении ГЛТЛ. Длительная терапия ИФН-α и малыми дозами цитостатических препаратов, обеспечивающая постоянное противоопухолевое воздействие, показывает свое преимущество перед короткоимпульсной интенсивной терапией и позволяет добиться улучшения общего состояния, регрессии клинических симптомов, нормализации лабораторных показателей и может быть применена в качестве индукционной терапии у больных ГЛТЛ. Однако достижения продолжительной полной ремиссии при длительной терапии ИФН-α и малыми дозами цитостатических препаратов не отмечено. Пациентам молодого возраста при достижении первой клинико-гематологической ремиссии заболевания при наличии сиблинга и в отсутствие тяжелой соматической патологии показана алло-ТКМ.
Обсуждение
В данной работе обобщен десятилетний опыт по диагностике и лечению больных ГЛТЛ в ФГБУ ГГЦ Минздрава России. Несмотря на то что ГЛТЛ отграничена как самостоятельная нозологическая единица более 20 лет назад, диагностика до сих пор представляет трудности, а оптимальная терапия не разработана. Большинство больных поступали с различными направительными диагнозами, что обусловлено наличием у больных ГЛТЛ неспецифичных клинических симптомов и лабораторных признаков, наблюдающихся при других гематологических заболеваниях и реактивных состояниях. В процессе обследования и верификации диагноза ГЛТЛ проводилась дифференциальная диагностика с хроническим Т-клеточным лейкозом из больших гранулярных лимфоцитов [29], миелодиспластическим синдромом, волосатоклеточным лейкозом [30], В-клеточной лимфомой селезенки [31, 32]. При установлении линейной принадлежности опухолевых клеток дифференциальная диагностика с МДС, ВКЛ, В-клеточными лимфомами селезенки не представляла особых трудностей. Наиболее сложно разграничить хронический Т-клеточный лейкоз из БГЛ и позитивные по CD8 варианты ГЛТЛ. Ранее ряд исследователей относил позитивные по CD8 случаи ГЛТЛ к хроническому Т-клеточному лейкозу из БГЛ, но отмечали отсутствие крупных азурофильных гранул в опухолевых клетках, большие размеры селезенки, более агрессивное клиническое течение [29]. В зарубежной литературе представлены описания случаев Т-клеточной лимфомы, занимающей по своим клинико-морфологическим характеристикам промежуточное положение между хроническим Т-клеточным лейкозом из БГЛ и ГЛТЛ. Заболевание манифестировало с В-симптомов, выраженной гепатоспленомегалии, лейкемизации опухолевого процесса. Авторами отмечена низкая эффективность цитостатического лечения [33].
Некоторые случаи позитивной по CD8 ГЛТЛ необходимо дифференцировать от реактивного лимфоцитоза, наблюдающегося при активной вирусной инфекции, в частности при реактивации эндогенных герпесвирусов. Комплексное клиническое обследование, включающее тестирование на расширенный спектр вирусных маркеров, позволяет в этих случаях провести дифференциальную диагностику между опухолевым и реактивным процессами либо констатировать их сочетание [34]. Ассоциация ГЛГЛ с реактивацией вирусной инфекции наблюдается редко, в литературе описаны единичные случаи выявления реактивации вирусной инфекции в дебюте ГЛТЛ преимущественно у больных с иммуносупрессией в анамнезе [35].
В большинстве представленных нами случаев ГЛТЛ мы наблюдали иммунофенотип CD3 + CD4 – CD8 – опухолевых клеток, в 3 случаях выявлены опухолевые клоны CD3 + CD4 + CD8 – , а в 6 случаях — CD3 + CD4 – CD8 + . По представленным клинико-морфологическим данным 15 больных ГЛТЛ из MD Anderson Cancer Center у 3 наблюдалась экспрессия антигена CD4, а у 2 иммунофенотип CD4 + CD8 + опухолевых клеток [7]. Представленные нами иммунофенотипические характеристики ГЛТЛ свидетельствуют о гетерогенности этой Т-клеточной лимфомы.
Считается, что ГЛТЛ характеризуется агрессивным клиническим течением, однако при ИГХИ биоптатов селезенки уровень Ki-67, отражающий пролиферативный потенциал, варьировал от 5 до 30%. Следует отметить выявления у 6 больных ЖКБ. По всей вероятности это тенденция, а не случайное совпадение: у этих 6 пациентов выявлены признаки внутрисосудистого гемолиза с гипербилирубинемией за счет непрямой фракции. Мы полагаем, что развитие ЖКБ является следствием длительно персистирующего гемолиза. Учитывая изложенное, низкий лимфопролиферативный потенциал, сопутствующую ЖКБ, значительные размеры селезенки, можно предположить, что некоторые случаи ГЛТЛ имеют индолентное течение. Клиническая симптоматика появляется при накоплении большой опухолевой массы, прогрессировании явлений гиперспленизма, появления иммунных нарушений, сопровождающихся гемолитической анемией.
ПЦР-диагностика клональных реаранжировок генов TCRD является важным маркером при диагностике ГЛТЛ, так как определяет степень зрелости опухолевых клеток. При созревании Т-лимфоцита в норме реаранжировки генов цепей TCR проходят последовательно: сначала перестраивается локус генов TCRD, затем проходят реаранжировки генов TCRG и неполные реаранжировки генов TCRB (Dβ—Jβ), а уже несколько позже происходят полные реаранжировки генов TCRB (Vβ—Jβ) и реаранжировки генов локуса α TCR (Vα—Jα) [23]. Так как гены TCRD располагаются внутри локуса α TCR, то они вырезаются при реаранжировках генов цепи α TCR. Спектр наблюдаемых при лимфомах клональных реаранжировок можно разделить на ранние (РР), свойственные γδ Т-клеточным лимфомам и более зрелые (ЗР), характерные для αβ Т-клеточных лимфом. Если в опухоли обнаруживают клональные продукты локусов TCRD, TCRG, неполные Dβ—Jβ реаранжировки, и отсутствуют полные Vβ—Jβ реаранжировки генов TCRB, то это свидетельствует о раннем характере реаранжировок. Среди обследованных 16 пациентов у 9 обнаружен ранний спектр реаранжировок (либо имелась клональная реаранжировка генов TCRD, либо отсутствовала полная реаранжировка генов TCRB) (см. табл. 3). По данным ИГХИ и ИФИ вариант γδ ГЛТЛ подтвержден у 4, а вариант αβ — у 8 больных. Однако в 3 случаях (больные № 3, 8, 18) наблюдали несоответствие данных иммунофенотипирования и молекулярных методов, т. е. при ИГХИ диагностирован вариант αβ опухоли в селезенке, а молекулярное исследование выявило ранние реаранжировки. Возможно, минорный клон клеток γδ не определяется при ИГХИ на фоне большого количества лимфоцитов αβ. Гены TCRD расположены в одном локусе с генами цепи α TCR. В процессе созревания лимфоцитов происходит вырезание генов цепи δ и T-лимфоциты αβ не несут перестроек генов TCRD. Поэтому с помощью молекулярного метода мы с высокой чувствительностью (10 –3 , т. е. 1 клональная Т-клетка γδ на 1000 Т-лимфоцитов αβ) можем обнаружить даже минорный Т-клеточный клон γδ (см. табл. 3).
Молекулярное исследование реаранжировок генов TCRD, TCRG и TCRB является важным звеном в диагностике ГЛТЛ, оно позволяет верифицировать вариант заболевания, распространенность опухолевого процесса, а также осуществлять оценку минимальной резидуальной болезни во время лечения.
Необходимость ИФИ клеток КМ, периферической крови, селезенки с помощью проточной цитометрии не вызывает сомнений. Оно позволяет комплексно исследовать иммунофенотипические особенности опухолевого клона, верифицировать вовлечение КМ, выявлять лейкемизацию опухолевого процесса.
Заключение
ГЛТЛ — гетерогенная группа заболеваний, обусловливающая необходимость комплексного обследования для дифференциальной диагностики с другими гематологическими заболеваниями и реактивными состояниями, протекающими со сходными клинико-лабораторными симптомами.
В соответствии с собственным опытом и данными зарубежных коллег нам представляется целесообразным проводить до начала противоопухолевого воздействия лечебно-диагностическую СЭ, позволяющую уменьшить объем опухолевой массы, восстановить показатели гемограммы, купировать иммунные нарушения и улучшить соматический статус больного. В качестве индукционной терапии может быть применена длительная терапия ИФН-α, малыми дозами цитостатических препаратов. Молодым пациентам, достигшим первой полной ремиссии в отсутствие тяжелой сопутствующей патологии, должна быть рекомендована высокодозная химиотерапия с трансплантацией аутологичных гемопоэтических клеток, а при наличии сиблинга — алло-ТГСК.
Хронический лимфоцитарный лейкоз

Хронический лимфоцитарный лейкоз – это постоянная выработка костным мозгом и накопление в тканях измененных лимфоцитов – клеток иммунной системы, которые не способны бороться с инфекциями и защищать организм от различных заболеваний.
Как развивается лимфолейкоз?
С самого рождения в теле человека, в том числе в костном мозге, образуются не только обычные клетки, но и отличающиеся от них мутировавшие – измененные. Они становятся такими из-за воздействия различных химических веществ, наследственных особенностей или случайных сбоев. Наш иммунитет выявляет и уничтожает большинство из них, но некоторым все же удается выжить. Они становятся лейкемическими и приобретают опасные свойства:
- перестают созревать и производить нормальные клетки крови;
- быстро размножаются;
- игнорируют апоптоз – механизм «запрограммированной» смерти, из-за чего существуют гораздо дольше, чем длится их правильный жизненный цикл.
Такие способности позволяют им накапливаться в костном мозге, вытеснять нормальные клетки, попадать в кровеносную систему, с ее помощью распространяться в различные области организма и препятствовать нормальной работе тканей.
При хроническом лейкозе такие клетки созревают не полностью – они становятся похожими на нормальные лейкоциты, необходимые нашему телу для борьбы с инфекциями, но не могут выполнять их функции.
Что такое костный мозг, кровь и лимфоидная ткань?
Различные типы лейкемии начинаются в определенных типах клеток крови. Для того, чтобы понять принцип развития заболевания и его влияния на организм, нужно разобраться с работой основных тканей тела.
Костный мозг
Эта мягкая внутренняя часть некоторых костей – черепа, лопаток, ребер, таза и позвоночника состоит из:
- Жировых клеток.
- Небольшого количества стволовых клеток, из которых образуются различные клетки крови.
- Зрелых кроветворных клеток.
- Поддерживающих тканей, которые помогают клеткам расти.
Внутри костного мозга стволовые клетки делятся, созревают и образуют новые клетки крови, которые становятся лейкоцитами, эритроцитами, лимфоцитами или тромбоцитами.

Типы клеток крови
Красные кровяные тельца, или эритроциты, переносят кислород из легких в другие ткани и возвращают углекислый газ обратно в дыхательные пути. Их нехватка – анемия, может вызвать слабость, усталость и одышку – нехватку воздуха.
Тромбоциты необходимы для закупорки вызванных порезами или ушибами отверстий в кровеносных сосудах. Если в теле человека их слишком мало, у него развиваются кровотечения и появляются кровоподтеки.
Белые кровяные тельца, или лейкоциты, помогают организму бороться с инфекциями. При понижении их уровня работа иммунной системы нарушается, а вероятность заражения – увеличивается.
Типы лейкоцитов
Лимфоциты – это полностью зрелые противостоящие инфекциям клетки, которые развиваются из лимфобластов – особого типа стволовых клеток костного мозга. Именно из них в основном состоит лимфоидная ткань иммунной системы, находящаяся в лимфатических узлах Лимфатические узлы – это небольшие органы иммунной системы, задерживающие и обезвреживающие опасные для организма вещества. , вилочковой железе Вилочковая железа, или тимус – небольшой орган, расположенных за верхней частью грудины, перед сердцем. В нем происходит созревание и развитие некоторых лимфоцитов. , селезенке Селезенка производит лимфоциты, уничтожает микроорганизмы и другие вредные вещества, а также хранит нормальные клетки крови. , миндалинах Миндалины – расположенные в задней части глотки скопления лимфатической ткани, которые участвуют в выработке антител – белков, не позволяющих вдыхаемым и проглатываемым микроорганизмам размножаться. , а также дыхательной и пищеварительной системах.
- В-лимфоциты: защищают организм от микробов. Именно они чаще всего становятся лейкозными. Созревают и превращаются в плазматические клетки, вырабатывающие антитела – белки, которые прикрепляются к микробам – бактериям, вирусам и грибкам, благодаря чему их распознают и уничтожают гранулоциты.
- Т-лимфоциты: распознают зараженные вирусами клетки, разрушают их и регулируют активность иммунной системы.
Гранулоциты: борются с инфекциями. Развиваются из особых клеток костного мозга – миелобластов, содержат в себе ферменты – белки и другие вещества, способные уничтожать бактерии.
Моноциты. Развиваются из присутствующих в костном мозге монобластов. На протяжении примерно суток они циркулируют в кровотоке, после чего проникают в различные ткани и превращаются в макрофаги, способные переваривать некоторые виды микробов. Кроме того, они помогают лимфоцитам распознавать опасные микроорганизмы и вырабатывать антитела для борьбы с ними.
Все эти клетки содержатся в теле человека в определенном количестве. Нарушение правильного баланса и замещение их на измененные клетки приводит к тяжелым последствиям – сбоям в работе всех систем, включая дыхательную, иммунную, кровеносную и пищеварительную, и отказу важнейших органов.
Типы лимфоцитарного лейкоза
Самый распространенный вид хронического лимфолейкоза развивается в В-лимфоцитах.
Пролимфоцитарный лейкоз: формируется в пролимфоцитах – незрелых В- или Т-лимфоцитах, и быстро распространяется. Большинство его случаев поддается лечению, но у обладателей подобного диагноза нередко случаются рецидивы – возвращение заболевания.
Лейкоз из больших гранулярных лимфоцитов. Его клетки довольно крупные и имеют признаки Т-, либо других типов лимфоцитов. Почти все такие лейкозы медленно развиваются и лечатся препаратами, подавляющими иммунную систему. Некоторые их формы довольно агрессивны – они быстро распространяются и плохо реагируют на терапию.
Волосатоклеточный лейкоз: формируется в измененных В-лимфоцитах. Его клетки необычно выглядят под микроскопом – на их поверхности есть тонкие выступы, похожие на волоски. Данное заболевание обычно хорошо поддается лечению.

Причины развития хронического лимфолейкоза
Точная причина большинства всех его случаев не известна, но принцип образования неправильных лимфоцитов понятен.
Нормальные человеческие клетки растут и работают по «программе», заложенной в содержащихся в них хромосомах, образующих молекулу ДНК. Она несет в себе переданные от родителей гены, которые влияют не только на наш внешний вид:
- онкогены: способствуют росту и делению клеток;
- супрессоры опухолей: замедляют их деление и вызывают гибель по окончанию жизненного цикла.
Каждый раз, когда клетка готовится к делению на 2 новых, она делает новую копию ДНК. Этот процесс не идеален – в его ходе могут возникать ошибки, приводящие к «включению» онкогенов и «выключению» супрессоров опухолей.
Нормальные В-лимфоциты являются частью нашей иммунной системы. Они «запрограммированы» на рост и деление с антигеном – чужеродным веществом, которое обычно не встречается в организме человека. К ним относятся, например, микробы – вирусы, бактерии и грибки. Хронический лимфолейкоз начинается в результате бесконтрольного деления В-лимфоцитов после их взаимодействия с антигеном. Причина этого медицинскому сообществу не понятна, но известны факторы, увеличивающие вероятность подобного события.
- Возраст: примерно 9 из 10 обладателей данного диагноза старше 50 лет.
- Некоторые исследования показали, что часть случаев связана с длительным воздействием радиоактивного газа радона и некоторых пестицидов – химических препаратов, которые используются для борьбы с сорняками и вредителями.
- Пол: у мужчин заболевание встречается несколько чаще, чем у женщин.
- Семейный анамнез: более чем в 2 раза повышены риски людей, у близких кровных родственников которых – родителей, братьев, сестер или детей, уже выявлено заболевание.
- Раса, этническая принадлежность. У проживающих в различных регионах и странах азиатов хронический лимфоцитарный лейкоз возникает реже, чем у европеоидов. Такая статистика подтверждает мнение ученых о том, что основную роль в его развитии играет генетика, а не окружающая среда.
Симптомы и признаки хронического лимфолейкоза
У многих пациентов на момент постановки диагноза никаких проблем со здоровьем нет. Заболевание часто обнаруживают по данным анализов крови при обращении к врачу из-за различных не связанных с ним нарушений.
Симптомы лимфолейкоза часто сложно распознать и легко принять за признаки различных расстройств. К ним относят:
- Слабость.
- Постоянную усталость.
- Увеличение размеров лимфатических узлов, похожих на плотные шишки под кожей.
- Потерю веса без усилий со стороны пациента.
- Повышенную температуру.
- Ночную потливость.
- Боль или чувство переполнения в области живота, сытость даже после небольшого приема пищи из-за увеличения селезенки или печени.
Многие симптомы заболевания возникают из-за замещения лейкозными клетками нормальных кроветворных клеток костного мозга. В результате этого процесса в организме пациентов присутствует слишком мало эритроцитов, правильно работающих лимфоцитов и тромбоцитов:
- Анемия – нехватка переносящих кислород эритроцитов. Приводит к слабости, усталости и одышке – ощущению нехватки воздуха.
- Лейкопения – дефицит нормальных лейкоцитов увеличивает риск развития инфекций.
- Нейтропения: низкий уровень нейтрофилов – особого типа гранулоцитов, необходимых для борьбы с бактериями.
- Лимфоцитоз – избыток измененных лимфоцитов, не способных бороться с инфекциями.
- Тромбоцитопения – нехватка тромбоцитов, приводящая к появлению большого количества кровоподтеков, частым или сильным носовым кровотечениям и кровоточивости десен.
У людей с хроническим лимфолейкозом из-за неправильной работы иммунитета развиваются инфекции – обычные простуда или герпес, пневмония – воспаление легких, или другие тяжелые заболевания.
Кроме того, в некоторых случаях лейкозные клетки вырабатывают неправильные антитела – белки, атакующие нормальные клетки крови. Эта особенность также приводит к снижению количества нормальных тромбоцитов, эритроцитов и лейкоцитов.

Диагностика хронического лимфолейкоза
Обследование начинается с опроса о беспокоящих симптомах, дате их появления, возможных факторах риска, общем самочувствии и состоянии здоровья, а также о известных тяжелых заболеваниях у кровных родственников. Затем проводится осмотр лимфатических узлов, брюшной полости и других областей тела, которые могли пострадать от лейкоза. После чего назначается целый ряд процедур.
- Анализы крови. Необходимы для подсчета различных типов клеток крови и химических веществ, а также качества работы внутренних органов, таких как печень и почки. У пациентов с хроническим лимфолейкозом очень много лимфоцитов – их содержание в количестве более чем 10 тысяч на кубический миллиметр крови фактически подтверждает диагноз.
- Анализ костного мозга. Как правило, для выявления заболевания достаточно данных показателей крови. Данное исследование проводится перед началом лечения для оценки его распространения и в процессе терапии для определения ее эффективности. Как правило, образцы берутся из задней части бедренной кости в ходе:
- аспирации – вытягивания густого жидкого костного мозга иглой в шприц;
- и обычно проводимой после нее биопсии – удаления небольшого кусочка кости и костного мозга с помощью специального инструмента.
Любые из вышеперечисленных исследований и диагностику лимфолейкоза можно пройти в онкологическом центре «Лапино-2».
Мы проводим полное обследование и берем все необходимые анализы, результаты которых получаем из собственной лаборатории.
Наши специалисты не только выявляют заболевания, но и проводят любое их лечение – без очередей и потерь времени.
Пациентам, проходящим терапию в других медицинских учреждениях, мы предлагаем услугу второго мнения – консультацию врача любой специальности. Независимые эксперты «Лапино-2» помогут вам убедиться в правильности уже поставленного диагноза или найти в нем ошибки и неточности.Стадии хронического лимфолейкоза
Для большинства видов онкологических заболеваний стадирование – это оценка количества поврежденных новообразованиями тканей. С помощью этой информации врачи определяют примерные прогнозы человека, и исходя из них подбирают самое подходящее лечение.
Хронический лимфоцитарный лейкоз же опухолей обычно не образует, но нередко к моменту выявления успевает распространить измененные клетки в другие ткани и органы. Перспективы его обладателей зависят от результатов лабораторных анализов и визуализирующих исследований.Лимфолейкоз стадируется по 2 международным системам, но в Российской Федерации применяется только одна из них – Binet. Она классифицирует заболевание по:
- количеству пораженных групп лимфоидной ткани, к которой относятся лимфоузлы шеи, паха и подмышек, селезенка и печень;
- наличию или отсутствию анемии – нехватки переносящих кислород эритроцитов, или
- тромбоцитопении – необходимых для правильной свертываемости крови тромбоцитов.
Стадия А: увеличено менее чем 3 участков лимфоидной ткани без анемии и тромбоцитопении.
Стадия В: укрупнено более чем 3 области лимфоидной ткани.
Стадия С: увеличено любое количество областей лимфоидной ткани, присутствуют анемия и/или тромбоцитопения.Лечение хронического лимфолейкоза
Многие пациенты с данным диагнозом живут очень долго, а доказательств пользы раннего начала лечения, тяжело переносимого организмом, на сегодняшний день не существует. Именно по этой причине врачи назначают его только после появления беспокоящих симптомов и прогрессирования, то есть ускорения развития заболевания.
В качестве начальной терапии специалисты применяют множество различных лекарств и их комбинаций. Они включают в себя:
- Моноклональные антитела – искусственные, то есть созданные в лаборатории белки иммунной системы, которые прикрепляются к определенной мишени – белкам, находящимся на поверхности измененных клеток.
- Химиотерапию – препараты, которые принимаются внутрь либо вводятся в вену или мышцу и уничтожают лейкозные клетки. Они попадают в кровоток и распространяются по всему телу, воздействуя на любые его области. Такое лечение проводится циклами, включающими в себя периоды отдыха, необходимые организму для восстановления.
- Таргетную терапию – препараты, которые работают только против определенных изменений, присутствующих в неправильных клетках. Они блокируют белки, которые помогают им быстро расти и существовать гораздо дольше, чем длится обычный жизненный цикл.
Если единственной серьезной проблемой является разрастание областей лимфоидной ткани, врачи назначают местное лечение:
- Лучевую терапию – разрушение лейкозных клеток с помощью радиации.
- Хирургию– удаление вызывающей тяжелые симптомы увеличенной селезенки.
В некоторых случаях избыток неправильных клеток приводит к лейкостазу – нарушению кровообращения. Их количество может уменьшить химиотерапия, но это происходит не раньше, чем через несколько дней после введения первой дозы препаратов. Перед ее проведением выполняется лейкаферез – изъятие излишков измененных клеток. Эта процедура сразу же улучшает показатели крови, но эффект от нее длится не долго.
Пациентам, у которых врачи обнаружили очень высокий риск развития заболевания, перед началом лечения может быть рекомендована трансплантация стволовых клеток, из которых образуются нормальные клетки крови.

Прогнозы и выживаемость при хроническом лимфолейкозе
Перспективы каждого пациента индивидуальны и зависят от огромного количества показателей.
Благоприятные прогностические факторы, связанные с длительной продолжительностью жизни:
- Не диффузное, то есть очаговое поражение костного мозга.
- Повреждение только одной части тринадцатой хромосомы.
- Низкая доля лейкозных клеток, содержащих менее 20% белка ZAP-70 или менее 30% CD38.
- Наличие клеток с измененным геном IGHV.
Неблагоприятные прогностические факторы, уменьшающие возможную продолжительность жизни:
- Пожилой возраст.
- Диффузное поражение костного мозга, то есть замещение большого его количества лейкозными клетками.
- Повреждение частей 17 или 11 хромосом.
- Трисомия – 3 двенадцатых хромосомы вместо двух в неправильных клетках.
- Лейкозные клетки без изменения гена IGHV.
- Высокий уровень в крови бета-2-микроглобулина – белка, присутствующего на поверхности почти всех клеток организма. Его количество в крови повышается при воспалениях, лимфоме, лейкозе и других заболеваниях.
- Отсутствие в измененных клетках гена TP53.
- Время, необходимое для удвоения количества лимфоцитов в организме – менее 1 года.
- Высокая доля лейкозных клеток, содержащих более 20% белка ZAP-70 или более 30% CD38.
- Избыток пролимфоцитов, из которых развиваются лимфоциты.
Продолжительность жизни пациентов с хроническим лимфолейкозом в последние годы значительно увеличилась. На сегодняшний день пятилетняя выживаемость, или вероятность прожить 5 или более лет с момента постановки диагноза, составляет 83%.
Зейналова Первин Айдыновна Гематолог, онколог, доктор медицинских наук, профессор, врач высшей категории. Заведующая отделением онкогематологии
Хронический лимфолейкоз
Хронический лимфолейкоз — злокачественное лимфопролиферативное заболевание, при котором опухолевыми клетками являются патологические В-лимфоциты, способные накапливаться в костном мозге, периферической крови и лимфатических узлах. В норме В-лимфоциты в течение своей жизни превращаются в иммуноглобулинсекретирующую клетку, которая обеспечивает приобретённый иммунитет. Опухолевые В-лимфоциты такой функции лишены, и, таким образом, иммунитет больного страдает, и повышается риск присоединения инфекционных заболеваний. Помимо этого, по мере прогрессирования заболевания нарушается выработка эритроцитов, нейтрофилов и тромбоцитов, возможно развитие аутоиммунных процессов. Наконец, хронический лимфолейкоз может трансформироваться в В-клеточный пролимфоцитарный лейкоз, в высокодифференцированную неходжкинскую лимфому, в частности — в диффузную В-крупноклеточную лимфому.
Причины развития лимфолейкоза
Хронический лейкоз является самым распространённым видом лейкоза, составляя до 30% в общей структуре заболеваемости. Частота встречаемости составляет 4 случая на 100 тысяч населения; у лиц старше 80 лет частота составляет более 30 случаев на 100 тысяч населения.
Факторами риска развития хронического лимфолейкоза являются:
- Пожилой возраст. До 70% всех выявленных случаев приходится на людей старше 60 лет,
- Мужской пол. Мужчины заболевают в два раза чаще женщин,
- Воздействие ионизирующего излучения,
- Контакт с бензолом и бензином.
Симптомы
Для хронического лимфолейкоза характерно длительное бессимптомное течение; основной причиной обращения к врачу являются изменения в общем анализе крови, сданном в рамках профилактического осмотра или по поводу иного заболевания. Активных жалоб на момент первичного осмотра пациент может и не предъявлять, но при этом зачастую даже в этой ситуации уже выявляется увеличение лимфатических узлов и изменение их консистенции до тестоватой. Сами лимфоузлы не уплотнены, сохраняют подвижность относительно окружающих тканей. В случае присоединения инфекции лимфатические узлы значительно увеличиваются; по мере прогрессирования хронического лимфолейкоза лимфоузлы — в первую очередь брюшной полости — способны
образовывать конгломераты.Первые возникающие жалобы обычно не носят специфического характера: повышенная утомляемость, слабость, выраженная потливость. По мере развития заболевания могут возникнуть аутоиммунные проявления, в первую очень гемолитические анемии (в 10-25% случаев) и тромбоцитопении (в 2-3% случаев). Гемолитическая анемия развивается в связи с разрушением самим организмом эритроцитов; чаще всего развивается, как и сам хронический лимфолейкоз, постепенно, но может проявиться и острым кризом — с повышением температуры, появлением желтухи, потемнения мочи. Тромбоцитопения может быть гораздо более опасным состоянием в связи с развитием кровотечений, в том числе и жизнеугрожающих (например, кровоизлияния в головной мозг).
Кроме того, так как В-лимфоциты относятся к клеткам, обеспечивающим иммунитет, типичным является присоединение инфекционных осложнений, в том числе оппортунистических, то есть вызванных микроорганизмами, постоянно находящимися в человеческом организме и не проявляющими себя при адекватном иммунном ответе. Чаще всего оппортунистическими инфекциями поражаются лёгкие.
Диагностика хронического лимфолейкоза
Диагноз хронического лимфолейкоза может быть заподозрен при оценке результатов обычного клинического анализа крови — обращает на себя внимание увеличение абсолютного количества лимфоцитов и лейкоцитов. Основным диагностическим критерием является абсолютное количество лимфоцитов, превышающее 5×10 9 л и прогрессивно увеличивающееся по мере развития лимфолейкоза, достигая цифр 100-500×10 9 л. Важно обращать внимание не только на абсолютное число, — если в начале заболевания лимфоциты составляют до 60-70% от всего количества лейкоцитов, то при его дальнейшем развитии они могут составлять 95-99%. Другие показатели крови, такие, как гемоглобин и тромбоциты, могут быть в норме, но при прогрессировании заболевания может быть выявлено их снижение. Абсолютным критерием для установки диагноза «хронический лимфолейкоз» является выявление более 5000 клональных В-лимфоцитов в 1 мкл периферической крови.
В биохимическом анализе крови может быть выявлено снижение общего белка и количества иммуноглобулинов, но это характерно для более поздних стадий заболевания. Обязательным этапом в диагностическом поиске является трепанбиопсия костного мозга. При гистологическом исследовании полученного пунктата на ранних этапах заболевания так же, как и в общем анализе крови, обнаруживается небольшое содержание лимфоцитов (40–50%), но при высоком лейкоцитозе лимфоциты могут составлять 95–98% костномозговых элементов.
Так как изменения в костном мозге являются неспецифическими, окончательный диагноз хронического лимфолейкоза устанавливается на основании данных иммуногистохимического исследования. Характерный иммунофенотип хронического лимфолейкоза включает экспрессию антигенов CD19, CD5, CD20, CD23, также отмечается слабая экспрессия на поверхности клеток иммуноглобулинов IgM (нередко одновременно с IgD) и антигенов CD20 и CD22. Главным цитогенетическим маркером, непосредственно влияющим на выбор терапии, является делеция 17p. Желательно выполнять анализ, направленный на выявлении этой делеции, до начала лечения, так как её выявление приводит к изменению тактики ведения пациента. Помимо биопсии костного мозга, в случае значительного увеличения отдельных лимфатических узлов показана пункция и их с целью исключения лимфомы.
Из инструментальных методов диагностики проводятся рентгенография органов грудной клетки и УЗИ наиболее часто поражаемых групп лимфоузлов и органов брюшной полости – в первую очередь печени и селезёнки, так как именно эти органы чаще всего поражаются при хроническом лимфолейкозе.

Стадии заболевания
В настоящее время стадирование осуществляется согласно классификации, предложенной Binet:
- A — содержание гемоглобина более 100 гл, тромбоцитов — более 100×10 9 л, поражено менее трёх лимфатических областей (к ним относятся: шейные лимфоузлы, подмышечные лимфоузлы, паховые лимфоузлы, печень, селезёнка),
- B — содержание гемоглобина более 100 гл, тромбоцитов — более 100×10 9 л, поражено более трёх лимфатических областей,
- C — содержание гемоглобина менее 100 г или тромбоцитов — менее 100×10 9 л.
Помимо классификации по Binet, используется классификация по Rai, используемая преимущественно в США. Согласно ей, выделяют четыре стадии заболевания:
- 0 — клинические проявления включают в себя только повышение лимфоцитов более 15×10 9 в периферической крови и более 40% в костном мозге,
- I — повышено количество лимфоцитов, диагностируется увеличение лимфоузлов,
- II — повышено количество лимфоцитов, диагностируется увеличение печени иили селезёнки вне зависимости от увеличения лимфоузлов,
- III — наблюдается повышение количества лимфоцитов и снижение уровня гемоглобина менее 110 гл вне зависимости от увеличения селезёнки, печени и лимфоузлов,
- IV — наблюдается повышение количества лимфоцитов и снижение числа тромбоцитов менее 100×10 9 вне зависимости от уровня гемоглобина, увеличения органов и лимфоузлов.
0 стадия характеризуется благоприятным прогнозом, I и II — промежуточным, III и IV — неблагоприятным.
Лечение
В настоящее время хронический лимфолейкоз хорошо поддаётся лечению благодаря широкому спектру химиотерапевтических препаратов. Важно отметить, что современные руководства не рекомендуют начинать агрессивное лечение сразу же после установки диагноза — в случаях, когда клинические проявления минимальны, возможно активное динамическое наблюдение до момента возникновения показаний к проведению специфического лечения, к коим относятся:
- Возникновение или нарастание интоксикации, которая проявляется потерей массы тела более чем на 10% за полгода, ухудшением общего состояния; появление лихорадки, субфебрильной температуры, ночных потов.
- Нарастание явлений анемии и/или тромбоцитопении;
- Аутоиммунная анемия и/или тромбоцитопения — в случае, если состояние не корректируется преднизолоном;
- Значительные размеры селезёнки — нижний край на расстоянии >6 см и более ниже рёберной дуги;
- Размер поражённых лимфатических узлов более 10 см или его прогрессивное увеличение;
- Увеличение количества лимфоцитов более, чем на 50% за 2 месяца, или вдвое за 6 месяцев.
Основным методом лечения хронического лимфолейкоза на данный момент является химиотерапия. Один из первых химиотерапевтических агентов, показавших свою эффективность в лечении хронического лимфолейкоза, хлорамбуцил, используется и в настоящее время, хоть и ограниченно. С течением времени вместо хлорамбуцила стали использовать циклофосфамид, в комбинации с другими препаратами, и соответствующие схемы (например, CHOP, COP, CAP) на данный момент применяются у пациентов молодого возраста с хорошим соматическим статусом.
Впервые введённый в клиническую практику в 80-х годах прошлого века флударабин показал эффективность в отношении достижения стойкой ремиссии, превышающую эффективность хлорамбуцила, особенно в сочетании с циклофосфамидом. Важно отметить, что эта схема эффективна даже в случае развития рецидива заболевания. Последним словом в лечении хронического лейкоза в настоящее время является применение иммунотерапевтических средств — препаратов из группы моноклональных тел. В рутинную клиническую практику прочно вошел ритуксимаб. Данный препарат взаимодействует с антигеном CD20, который ограниченно экспрессируется при хроническом лимфолейкозе, поэтому для эффективного лечения требуется сочетание ритуксимаба с какой-либо из принятых схем химиотерапии, чаще всего с флударабином или COP. Ритуксимаб в монорежиме может применяться как поддерживающая терапия при частичном ответе на лечение.
Перспективным выглядит применение препарата алемтузумаб, который взаимодействует с антигеном CD52. Его также используют как в монорежиме, так и в комбинации с флударабином.
Отдельно хотелось бы упомянуть хронический лимфолейкоз с делецией 17p. Этот подтип лимфолейкоза часто бывает резистентен к стандартным схемам химиотерапии.
Определённые успехи в лечении этого подвида лимфолейкоза достигнуты благодаря применению упомянутого выше алемтузумаба. Кроме того, перспективным средством в этой ситуации является ибрутиниб. В настоящее время этот препарат применяется в монорежиме, сочетание его с различными схемами химиотерапии исследуется; определённое преимущество показала схема, включающая ибрутиниб, ещё одно моноклональное тело — ритуксимаб, и бендамустин.

Лучевая терапия, которая столетие назад была практически единственной возможностью лечения таких больных, и по сей день не утратила актуальности: рекомендуется её проведение в рамках комплексного подхода на область поражённых лимфоузлов, если наблюдается их продолженный рост на фоне стабилизации остальных проявлений заболевания. В этом случае необходимая суммарная доза составляет 20-30 Гр. Также лучевой метод может быть применён при рецидивах заболевания.
В лечении хронического лимфолейкоза нашёл своё место и хирургический метод, заключающийся в удалении поражённой селезёнки. Показаниями к данному вмешательству являются:
- Увеличение селезёнки в сочетании с тяжелой анемией и/или тромбоцитопенией, особенно если наблюдается химиорезистентность,
- Массивное увеличение селезёнки при условии отсутствия ответа на химиотерапию,
- Тяжёлая аутоиммунная анемия и/или тромбоцитопения при резистентности к медикаментозному лечению.
При развитии резистентности к применяемым ранее химиотерапевтическим агентам или же при быстром прогрессировании после проведённого лечения может быть проведена трансплантация костного мозга. Трансплантация костного мозга показана в первой ремиссии пациентам из группы высокого риска, молодым пациентам в отсутствие эффекта от проводимого лечения, пациентам с делецией 17p/мутацией TP53 при наличии прогрессии заболевания. Важно отметить, что после проведённой трансплантации рекомендуется применение ритуксимаба и леналидомида в качестве поддерживающей терапии с целью предотвращения рецидива.
Наконец, пациентам требуется проведение и поддерживающей терапии, которая включает:
- Переливание эритроцитарной массы при анемии;
- Переливание тромбоцитарной массы при кровотечении, вызванном тромбоцитопенией;
- Противомикробные средства при присоединении бактериальной, грибковой или вирусной инфекции, а также для её профилактики;
- Применение преднизолона в дозе 1-2 мг/кг при развитии аутоиммунных процессов.
В случае развития рецидива заболевания тактика лечения зависит от ряда факторов, таких как: проведённая ранее терапия, срок наступления рецидива, клиническая картина. В случае раннего (то есть возникшего в периоде 24 месяцев и ранее) рецидива основным препаратом является ибрутиниб. Он применяется как самостоятельно, так и в составе упомянутой выше схемы лечения (ибрутиниб+ритуксимаб+бендамустин).
Альтернативным препаратом выбора может быть алемтузумаб. Демонстрируя сопоставимую с ибрутинибом эффективность, он, однако, демонстрирует значительно большую токсичность.
Наконец, у ряда пациентов по поводу раннего рецидива хронического лимфолейкоза может быть выполнена трансплантация костного мозга.
В случае позднего (возникшего в срок более 24 месяцев с момента завершения лечения) рецидива основным критерием выбора является проведённая ранняя терапия.
- Если проводимая ранее терапия на основе флударабина не сопровождалась значительной токсичностью, то можно вернуться к этой схеме, а также дополнить её ритуксимабом.
- В случае выявления цитопении возможно применение ритуксимаба в сочетании с высокими дозами глюкокортикостероидов.
- При проведённом ранее лечении хлорамбуцилом показано применение схем с флударабином или сочетанием бендамустина и ритуксимаба.
- Монотерапия ибрутинибом или его сочетание с одной из схем полихимиотерапии также может быть эффективна при рецидиве хронического лимфолейкоза.
Оценка эффективности лечения
Диагностические исследования, направленные на оценку эффекта от проведённого лечения, проводятся не ранее, чем через 2 месяца после окончания последнего курса химиотерапии. Результат может быть оценён следующим образом:
- Полная ремиссия: уменьшение до нормальных размеров печени, селезёнки, лимфоузлов (допустимо их увеличение в размере не более чем 1,5 см), снижение числа лимфоцитов менее 4×10 9 л в периферической крови и менее 30% в костном мозге, повышение числа тромбоцитов более 100×10 9 л, гемоглобина — более 110 гл, нейтрофилов — более 1,54×10 9 л.
- Частичная ремиссия: уменьшение размеров лимфоузлов, печени и селезёнки на 50% и более, снижение количества лимфоцитов в периферической крови на 50%, повышение числа тромбоцитов более 100×10 9 л, гемоглобина — более 110 гл, нейтрофилов — более 1,54×10 9 л или же повышение любого из этих параметров более чем на 50% от исходного уровня.
- Признаками прогрессирования заболевания являются, напротив, увеличение размеров лимфоузлов, печени и селезёнки на 50% и более, а также уменьшение количества тромбоцитов на 50% и более от исходного уровня и уменьшение количества тромбоцитов на 20 гл и более.
Для установления полной ремиссии необходимо соблюдение всех перечисленных критериев, частичной — по крайней мере 2 критерия, касающихся состояния внутренних органов, и минимум одного критерия, касающегося клеточного состава крови.
Следует учитывать, что терапия ибрутинибом может привести к полному ответу со стороны лимфатических узлов и селезёнки, но с сохранением лейкоцитоза в периферической крови. Это состояние обозначается, как частичный ответ с лимфоцитозом.
Прогноз
Значительные успехи в терапии хронического лимфолейкоза позволили сделать это заболевание потенциально излечимым или же достаточно долго поддерживать жизнь человека без прогрессирования основного заболевания с сохранением её качества.
Напротив, без лечения заболевание медленно, но неуклонно прогрессирует, что способно стать причиной гибели пациента спустя несколько лет после дебюта заболевания, поэтому своевременное обращение к врачу и начало адекватной терапии очень важны.
Источник https://www.mediasphera.ru/issues/terapevticheskij-arkhiv/2016/7/1004036602016071004
Источник https://lapino2.ru/napravleniya/onkogematologiya/khronicheskiy-limfoleykoz/
Источник https://www.euroonco.ru/onkogematologiya/hronicheskij-limfolejkoz