Фенилкетонурия
Фенилкетонурия (ФКУ) – это врожденная аутосомно-рецессивное заболевание из группы ферментопатий (заболеваний, при которых нарушается работа ферментов), связанное с нарушением обмена аминокислоты фенилаланина. Фенилаланин является незаменимой аминокислотой, поступающей в организм человека с белковой пищей. В организме человека фенилаланин используется для синтеза белков, а неиспользованный запас превращается в аминокислоту тирозин. При фенилкетонурии обмен аминокислоты нарушается, и ее уровень начинает повышаться.
Заболевание встречается у 1 ребенка на 10 000 новорожденных с одинаковой частотой среди мальчиков и девочек.

Суперфуды в косметике: сочные коктейли для здоровья кожи и волос
Симптомы
Клиническая картина фенилкетонурии развивается в первом полугодии жизни и с ростом малыша прогрессирует. На первых месяцах жизни ребенок становится вялым, либо появляются раздражительность, плаксивость, беспокойство, отсутствие интереса к окружающему миру, игрушкам. Он отстает в психическом, умственном развитии, замедляется рост черепа. Для таких детей характерен специфический затхлый «мышиный» запах тела.
Основные предъявляемые жалобы (со слов родителей): при отсутствии лечения на первом году жизни, обычно в возрасте 2−6 месяцев, родителей беспокоят срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония – слабость мышц), признаки атопического дерматита, задержка психомоторного развития, иногда судороги.
Ребенок поздно учится сидеть, ползать, ходить, зубы прорезываются позже, чем у сверстников. Формируется специфическая поза и осанка при сидении. Нередко могут присоединиться симптомы поражения центральной нервной системы: навязчивые состояния, судороги, тремор (дрожание конечностей) с дальнейшим формированием эпилепсии. Как правило, дети голубоглазые, светловолосые, кожа светлая (содержит мало пигмента меланина), сухая с шелушением. У темнокожих и темноволосых детей, заболевание встречается очень редко.
Формы
Классификация классической фенилкетонурии по форме заболевания исходя из содержания фенилаланин в крови: легкая гиперфенилаланинемия (не ФКУ) — содержание фенилаланина 120−600 мкмоль/л; умеренная (мягкая, средняя) ФКУ — содержание фенилаланина 600−1200 мкмоль/л; классическая (тяжелая) ФКУ, когда уровень фенилаланин превышает 1200 мкмоль/л.
Причины
Основной причиной развития данной патологии является отсутствие или очень низкая выработка печеночного фермента фенилаланингидроксилазы, который превращает фермент фенилаланин в тирозин. В результате отсутствия данного фермента в организме начинает накапливаться фенилаланин и его производные, которые оказывают токсическое воздействие на организм ребенка, в частности, на центральную нервную систему.
Методы диагностики
Диагностика фенилкетонурии осуществляется врачом-педиатром на основании сбора жалоб, данных анамнеза, клинического осмотра, лабораторных (включая генетические) и дополнительных инструментальных методов обследования. Диагностика направлена на определение клинической формы заболевания, его причины, тяжести состояния и возникающих осложнений.
Все новорожденные дети уже в роддоме на 3-4 сутки (недоношенным на 7-е сутки) по программе неонатального скрининга обследуются на фенилкетонурию. Содержание фенилаланина выше 2,0 мг/дл классифицируется как гиперфенилаланинемия, которая требует проведения уточняющей диагностики. Определяют концентрацию фенилаланина в сухих пятнах крови новорожденных методом флуориметрии (метод определения концентрации вещества по интенсивности флуоресценции, возникающей при облучении вещества монохроматическим излучением) и методом тандемной масс-спектрометрии (метод исследования вещества, основанный на определении отношения массы к заряду ионов,).
В настоящее время известно более 900 мутаций в гене РАН, которые могут привести к развитию фенилкетонурии: поэтому показано генетическое исследование. В биохимическом анализе крови и мочи может определяться снижение содержания гомованилиновой кислоты и 5-оксииндолуксусной кислоты, содержание кетоновых тел повышено.
Для выявления изменений в различных органах и наличии осложнений назначают проведение инструментальных методов исследования – электроэнцефалография, МРТ – магнитно-резонансная томография с целью выявления очагов поражения коры мозга и других изменений у пациентов старше 12 лет; УЗИ брюшной полости и почек для диагностики дискинезии (нарушения моторики) желчных путей, диффузных изменений печени и поджелудочной железы, мочекаменной болезни; фиброгастродуоденоскопия (проводится по показаниям) – для диагностики поражения слизистой оболочки желудка.
Дифференциальная диагностика фенилкетонурии проводится с транзиторной гиперфенилаланинемией недоношенных, тирозинемией (нарушения метаболизма с повреждением печени, почек, периферических нервов), галактоземией (наследственное заболевание, в основе которого лежит нарушение обмена веществ на пути преобразования галактозы в глюкозу), другими заболеваниями, связанными с нарушением функции печени.
Основные используемые лабораторные исследования:
- Определение концентраций фенилаланина в сухих пятнах крови методом флуориметрии (в первые дни жизни).
- Фенилаланин в крови (тандемная масс-спектрометрия) – увеличение;
- Проба Фелинга (определение фенилпировиноградной кислоты в моче) – положительна.
- Генетическое исследование мутации гена PAH: мутация R408W, мутации IVS12nt1, R261Q, R252W, R158Q, P281L, IVS10nt546, I65T (предрасположенность к развитию фенилкетонурии).
Дополнительные используемые лабораторные исследования:
- Птерины в моче: снижение образования белка биоптерина (кофермент, участвующий в ряде важных биохимических реакций) при мутации гена PTS; при мутации в гене DHPR – образование биоптерина повышено.
- Кетоновые кислоты в моче повышены.
- Гомованилиновая кислота, 5-оксииндолуксусной кислота (в крови) повышены.
- Генетическое исследование на гиперфенилаланемию – гены: PAH, GCH1, DHPR, PCBD1, PTS, QDPR.
Основные используемые инструментальные исследования:
- Электроэнцефалография;
- МРТ головного мозга (очаги повреждения коры головного мозга);
- УЗИ органов брюшной полости;
- Эзофагогастроскопия (диагностика поражения слизистой желудка).
Лечение
Лечение фенилкетонурии заключается в — снижении фенилаланина в крови, повышении толерантности (переносимость) фенилаланина, получаемого с натуральной пищей. И таким образом избежать тяжелой неврологической симптоматики и улучшить качество жизни.
Диетотерапия с исключением продуктов содержащих фенилаланин (рыбные, молочные, мясные продукты). Категорически запрещается использование газированных напитков, содержащих фенилаланин. Для вскармливания грудных детей разработаны специальные смеси с низким содержанием фенилаланина. Своевременное выявление патологии и соблюдение диеты позволяет избежать развитие необратимых процессов со стороны центральной нервной системы.
Все дети с фенилкетонурией подлежат наблюдению участковым педиатром и психоневрологом с проведением контроля уровня в крови фенилаланина.
Осложнения
Возможны развитие микроцефалия (маленький размер черепа), задержка умственного развития или умственная отсталость, олигофрения.
Профилактика
Профилактика фенилкетонурии включает несколько мер:
- Медико-генетическое консультирование пар с рекомендацией обследования на гетерозиготное носительство частых мутаций в гене РАН;
- Неонатальный скрининг: лабораторное обследование для своевременного выявления и начала лечения больных детей с целью предотвращения их инвалидизации;
- Пренатальная диагностика фенилкетонурии в семье, где уже есть ребенок с фенилкетонурией (выявления фенилкетонурии на стадии внутриутробного развития путем определения активности фенилаланингидроксилазы в культуре амниоцитов (клеток плодного пузыря);
- Профилактика рождения детей с синдромом «материнской фенилкетонурии» от женщин, больных ФКУ, путем организации психологической помощи девочкам-подросткам по вопросам необходимости соблюдения строгой гипофенилаланиновой диеты в пубертатный период, а также консультативной помощи по вопросам планирования семьи и беременности.
Какие вопросы следует задать врачу
Влияет ли фенилкетонурия на развитие детей?
Как можно выявить фенилкетонурию?
Существует ли лечение фенилкетонурии?
Советы пациенту
Поскольку фенилаланин составляет 4-6% всех пищевых белков, продукты с высоким содержанием белка, такие как мясо, рыба, яйца, сыр, орехи, соя, как правило, не разрешается употреблять пациентам с высокой степенью тяжести фенилкетонурии. Потребность в белках и азоте обеспечивается при помощи белковых заменителей, в которых не содержится фенилаланин. Необходимо обеспечение организма всеми витаминами, минералами, незаменимыми жирными кислотами и ненасыщенными жирными кислотами с длинной цепью для поддержания потребности в питательных веществах.
Фенилкетонурия — симптомы и лечение
Что такое фенилкетонурия? Причины возникновения, диагностику и методы лечения разберем в статье доктора Алексенцевой Елены Сергеевны, педиатра со стажем в 14 лет.
Над статьей доктора Алексенцевой Елены Сергеевны работали литературный редактор Юлия Липовская , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина

Определение болезни. Причины заболевания
Фенилкетонурия (ФКУ) — генетическое заболевание, в основе которого лежит врождённое нарушение метаболизма аминокислот, характеризующееся повышенным содержанием фенилаланина в крови. Это аутосомно-рецессивная патология, т. е. ребёнок может унаследовать данное заболевание только в том случае, если оба родителя являются носителями дефектной версии гена.
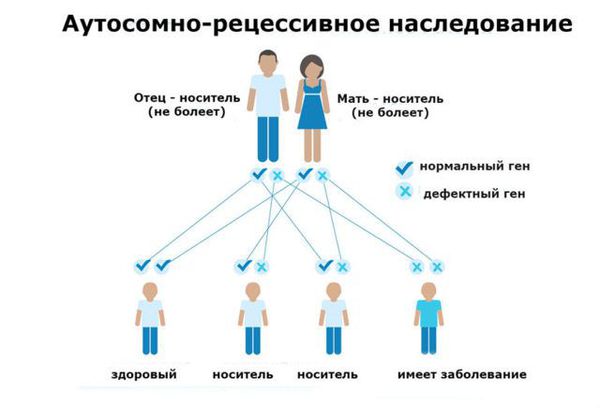
ФКУ связана с дефектом в гене PAH (Phenylalanine hydroxylase gene). Этот ген участвует в образовании фенилаланин-гидроксилазы — печёночного фермента, ответственного за расщепление фенилаланина. Из-за дефекта гена количество этого фермента снижается, а уровень фенилаланина в организме увеличивается, что влечёт за собой такие тяжёлые и необратимые последствия, как умственная отсталость глубокой степени и эпилептические приступы, плохо отвечающие на стандартную антиконвульсантную терапию. П ытаясь избавиться от избытка фенилаланина и его метаболитов ( фенилпировиноградной и фенилуксусной кислот), организм выводит их с мочой .
Распространённость фенилкетонурии
Распространённость фенилкетонурии широко варьируется во всём мире. В Европе частота встречаемости данного заболевания составляет 1 случай на 10 000 живорождённых детей, но для некоторых районов она значительно выше, что связано с большим количеством близкородственных браков. Например, в Турции фенилкетонурия выявляется примерно у одного из 4000 живорождённых детей [1] . В Финляндии, по данным статистики, зарегистрирована самая низкая частота ФКУ среди Европейских стран: 1 случай на 100 000 детей [2] . Что касается нашей страны, усреднённый показатель распространённости данного заболевания — 1 случай на 7000 живорождённых детей.
Факторы риска фенилкетонурии
Основной фактор риска фенилкетонурии — это наличие у обоих родителей дефекта в гене PAH (Phenylalanine hydroxylase gene). Заболевание развивается, если оба родителя передают ребёнку копию повреждённого гена.
Другим ф актором развития фенилкетонурии можно назвать этническую принадлежность ребёнка. Считается, что у афроамериканцев данное заболевание встречается реже, чем у других этнических групп [22] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы фенилкетонурии
Как проявляется фенилкетонурия у новорождённых
У новорождённых с фенилкетонурией симптомы изначально отсутствуют и развиваются медленно, в течение нескольких месяцев, что связанно с постепенным накоплением в организме фенилаланина [3] . Выявить заболевание можно только с помощью неонатального скрининга.
Чем дольше ребёнок с фенилкетонурией не получает специфического лечения, тем быстрее развивается умственная отсталость и необратимые нарушения развития. Кроме того, для детей с фенилкетонурией характерны следующие признаки [4] :
- специфический неприятный запах изо рта, от кожи или мочи, который можно сравнить с затхлым «мышиным» запахом;
- приступы тошноты и рвоты;
- светочувствительность;
- неврологические нарушения, которые нередко могут включать эпилепсию (50 %);
- экстрапирамидные нарушения (паркинсонизм — неврологический синдром, который характеризуется тремором в состоянии покоя, мышечной ригидностью и общей замедленностью движений);
- нарушения зрения: косоглазие, атрофия зрительного нерва, снижение остроты зрения;
- кожные высыпания по типу экземы, локализующиеся на любых участках тела;
- светлая кожа и голубые глаза (из-за невозможности фенилаланина превращаться в меланин);
- микроцефалия (уменьшение размеров черепа и головного мозга при нормальных размерах других частей тела);
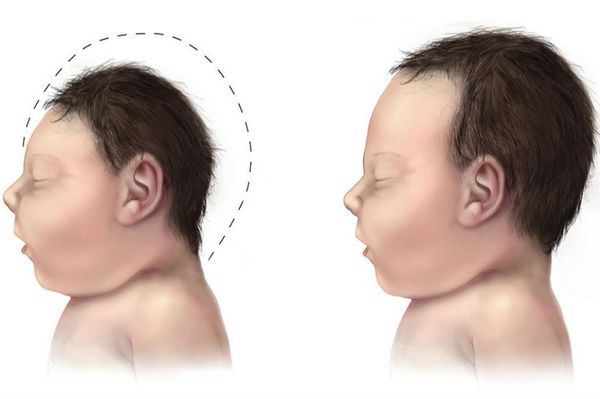
- гиперактивность: беспокойство, неконтролируемые вспышки гнева, невозможность сосредоточиться на выполняемых задачах, спонтанное снижение концентрации;
- интеллектуальная недееспособность: от лёгкой недостаточности интеллекта до идиотии и имбецильности;
- проблемы в поведенческой, эмоциональной и социальной сферах: трудности обучения в образовательных учреждениях, снижение мотивации при выполнении рабочих задач, снижение социальных коммуникаций, раздражительность, перепады настроения, социальная изоляция;
- психические расстройства: депрессии, генерализованные тревоги, шизофрения, биполярное расстройство.
У детей, рождённых от матерей с фенилкетонурией и неконтролируемым уровнем фенилаланина в крови, риск умственной отсталости значительно выше , поскольку они подвергаются воздействию очень высоких и токсичных уровней фенилаланина ещё до рождения. Такие дети могут иметь низкий вес при рождении, что вызывает проблемы прибавки массы и физического развития в будущем. Кроме того, умственно они развиваются значительно медленнее, чем их здоровые сверстники. Другими характерными признаками являются различные пороки сердечно-сосудистой системы (тетрада Фалло, коарктация аорты, дефект межжелудочковой перегородки, дефект межпредсердной перегородки) и поведенческие проблемы, связанные в будущем с нарушением социальной адаптации [5] .
Патогенез фенилкетонурии
Фенилкетонурия как самостоятельное заболевание было открыто норвежским врачом Иваром Асбьёрном Фёллингом ещё в 1934 году. Несмотря на это, вопрос о патогенезе долгое время оставался открытым.
Фенилаланин — это незаменимая аминокислота, которая участвует с синтезе белков. Незаменимая она потому, что организм не может самостоятельно её синтезировать, фенилаланин можно получить исключительно из пищи (мясных и рыбных продуктов, творога, сыра, яйц, орехов, хлебобулочных изделий, круп) или с помощью протеолиза — процесса гидролиза белков с помощью ферментов-протеаз.
Основной метаболический путь фенилаланина включает его преобразование до тирозина под действием фермента фенилаланин-гидроксилазы, который обнаруживается в печени и почках. Тирозин — это аминокислота, которая участвует в образовании таких важных соединений, как нейромедиаторы (дофамин, адреналин и норадреналин) и пигмент меланин. Нейромедиаторы передают электрические импульсы между нервными клетками (нейронами) и от нейронов к другим клеткам (например мышечным или железистым), что имеет немаловажную роль в когнитивной деятельности. Пигмент меланин защищает организм человека от воздействия ультрафиолетовых лучей [6] .
У пациентов, страдающих фенилкетонурией, из-за дефекта гена и недостатка фермента фенилаланин-гидроксилазы происходит увеличение в плазме крови концентрации фенилаланина (более 1200 мкмоль/л при норме 0-120 мкмоль/л ) и его метаболитов. Одновременно с этим снижается уровень тирозина и его производных (дофамина, адреналина, норадреналина и меланина). Такое состояние оказывает выраженное нейротоксическое действие на структуры мозга. Если пациент с фенилкетонурией не получает или не соблюдает лечение, то у него отмечаются повреждения мозолистого тела, полосатого тела, изменения коры и гипомиелинизация (снижение содержания миелина, образующего оболочку нервных волокон, в различных структурах оболочек мозга). Эти изменения могут привести к снижению интеллектуального развития и нейродегенерации — прогрессирующей гибели нервных клеток. Поэтому пациенты с фенилкетонурией более восприимчивы к нарушениям, связанным с дефицитом дофамина в головном мозге, таким как паркинсонизм.
Хотя патофизиологические механизмы повреждения головного мозга у пациентов с фенилкетонурией ещё не совсем понятны, существует множество свидетельств метаболических изменений, которые включают:
- дефицит биоэнергетики (нарушение процессов преобразования энергии в организме человека);
- окислительный стресс;
- нарушение метаболизма липидов и белков;
- нарушение оптимального уровня кальция и синтеза нейромедиаторов в головном мозге.
Окислительный стресс — это повреждение клеток активными формами кислорода, которые представляют собой молекулы с повышенной реактивностью из-за наличия неспаренного электрона на внешнем электронном уровне. Активные формы кислорода образуются в клетках постоянно, но в норме их уровень настолько низкий, что организм самостоятельно их нейтрализует с помощью антиоксидантной системы. Окислительный стресс происходит в том случае, когда активных форм кислорода образуется слишком много и антиоксиданты не могут полностью их инактивировать. Такой дисбаланс может вызвать окислительное повреждение белков, липидов или ДНК.
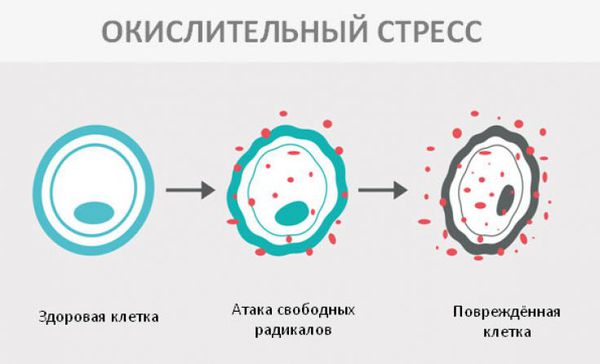
В ходе исследований окислительного повреждения макромолекул у пациентов с фенилкетонурией удалось установить, что увеличение концентрации фенилаланина в сыворотке крови (гиперфенилаланинемия) вызывает снижение антиоксидантоной защиты, что и приводит к окислительному стрессу [8] .
Ткани мозга особенно уязвимы для окислительного стресса из-за высокого потребления кислорода, высоких концентраций железа в тканях, низкого уровня антиоксидантной защиты, что способствует избыточному синтезу перекиси водорода (одной из форм активного кислорода) [7] .
Классификация и стадии развития фенилкетонурии
Согласно Европейской классификации, различают несколько вариантов течения данного заболевания [12] .
- Классическая фенилкетонурия характеризуется полным или практически полным дефицитом фенилаланин-гидроксилазы в печени. Сопровождается диетической переносимостью фенилаланина < 250-350 мг/день и концентрацией фенилаланина в крови при нормальной диете свыше 1200 мкмоль/л. При отсутствии своевременной диагностики первые клинические признаки заболевания отмечаются уже на первом году жизни ребёнка: родители жалуются на общую вялость, частые беспокойства, срыгивания при кормлении, проявления атопического дерматита. При осмотре ребёнка отмечается мышечная гипотония, задержка нервно-психического развития, в некоторых случаях — микроцефалия. Отличительной особенностью являются эпилептические приступы, плохо поддающиеся купированию стандартными схемами антиконвульсантной терапии.
- Умеренная фенилкетонурия характеризуется низкой остаточной ферментативной активностью (низкой способностью имеющихся ферментов участвовать в расщеплении фенилаланина и его превращении в тирозин), переносимостью фенилаланина от 350 до 400 мг/день и концентрацией фенилаланина в крови 600-1200 мкмоль/л. Данная форма отличается от классической фенилкетонурии более поздней манифестацией (чаще — первый-второй год жизни) и более медленным прогрессированием клинических признаков при отсутствии лечения.
- Лёгкая гиперфенилаланинемия. Уровень фенилаланина в плазме крови составляет менее 600 мкмоль/л при обычной диете. При данной форме у детей клинические признаки заболевания отсутствуют, либо незначительны. Несмотря на это, такие дети требуют особого контроля содержания фенилаланина в крови и непрерывной оценки нервно-психического развития.
Осложнения фенилкетонурии
Самым грозным осложнением фенилкетонурии является прогрессирующая умственная отсталость при отсутствии лечения. Однако данное заболевание ассоциируется также с рядом других патологий, влияющих на жизнь и здоровье пациента.
Доказано, что уровень депрессии среди взрослых с фенилкетонурией значительно выше и составляет около 18 % [10] . У них отмечается резкое снижение продуктивности, нарушения сна, выраженная утомляемость на фоне привычной деятельности, подавленное настроение, потеря интереса к жизни и привычным делам, нарушения аппетита, мысли о самоубийстве.
Также у взрослых пациентов с фенилкетонурией очень высока распространённость неврологических симптомов: тремор рук (присутствует у трети пациентов), генерализованная дистония (неритмичные медленные насильственные движения в различных частях тела, которые сопровождаются своеобразным изменением мышечного тонуса), задержка когнитивного и моторного развития (особенно при нарушениях диетотерапии), паркинсонизм [11] .
Пожизненная диетотерапия ассоциирована с нарушением роста, снижением минеральной плотностей костей и дефицитом питательных веществ, что требует постоянного контроля у профильных специалистов.
Диагностика фенилкетонурии
Первым эффективным тестом на определение у пациента гиперфенилаланинемии был бактериальный анализ ингибирования, разработанный американским микробиологом Робертом Гатри в 1963 году. Этот тест был основан на потребностях фенилаланина для роста культуры грамположительной бактерии Bacillus subtilis. Данный тест был очень удобен для массового скрининга, т. к. пятно засохшей крови можно было получить в кабинете врача, используя специальную фильтровальную бумагу, а затем отправить по почте в конверте в необходимую лабораторию [13] .
Развитие медицины привело к созданию тандемной масс-спектрометрии для быстрого определения концентраций аминокислот в небольших объёмах крови или плазмы. Данный метод даёт более низкую частоту ложноположительных результатов, измеряя уровни фенилаланина и тирозина в исследуемых образцах.
Основным методом ранней диагностики фенилкетонурии во всех странах мира принято считать неонатальный скрининг, который проводится в строго декретированные сроки для обеспечения своевременного начала лечения [12] . Неонатальный скрининг — это генетическое тестирование, которое позволяет выявить наиболее распространённые врождённые и наследственные заболевания у новорождённых детей.
В нашей стране проводится массовое обследование новорождённых детей на 5 наследственных заболеваний, одним из которых является ФКУ. Неонатальный скрининг проводится на четвёртые сутки жизни у доношенного ребёнка и на седьмые сутки у недоношенного ребёнка. В паспорте новорождённого и его истории болезни ставится пометка о проведении [14] .
Для неонатального скрининга медицинским персоналом с помощью скарификатора осуществляется забор крови из пятки новорождённого строго через 3 часа после кормления. Полученные образцы крови помещаются на специальные фильтровальные бумажные тест-бланки и отправляются в лабораторию.
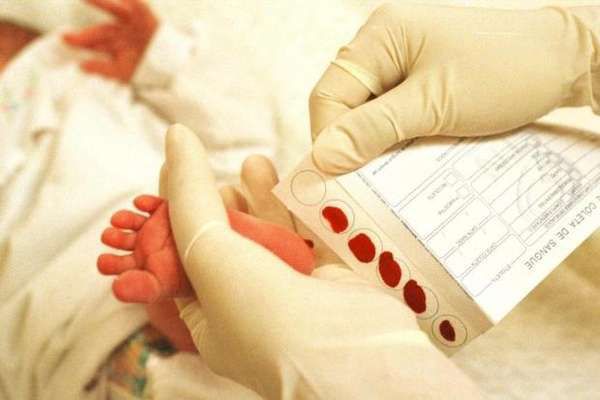
Стоит отметить, что у некоторых детей, особенно у рождённых раньше срока, может наблюдаться незрелость ферментных систем, участвующих в метаболизме аминокислот. Это приводит к кратковременному повышению фенилаланина и положительному результату при скрининге [12] Около 2 % всех случаев повышенного уровня фенилаланина в крови, выявленного при скрининге новорождённых, связаны с нарушением метаболизма кофермента BH4, который участвует в преобразовании фенилаланина. Это подчёркивает важность проведения дифференциальной диагностики при всех выявленных уровнях гиперфенилаланинемии. Фенилкеторурию необходимо дифференцировать с такими заболеваниями, как транзиторная гиперфенилаланинемия недоношенных, наследственная доброкачественная гиперфенилаланинемия, тирозинемия, галактоземия.
Генотипирование не является необходимым методом для диагностики фенилкетонурии, но помогает специалистам определять степень дисфункции белка, остаточную активность фенилаланин-гидроксилазы , и, следовательно, метаболический фенотип, на основании которого в дальнейшем будет строиться тактика лечения.
Лечение фенилкетонурии
В 1990 г. исследователь Smith и его коллеги в своей работе доказали, что отсрочка старта лечения фенилкетонурии на каждые четыре недели приводит к падению показателя коэффициента интеллекта (IQ) примерно на 4 балла [15] .
Последнее Европейское руководство по фенилкетонурии акцентирует внимание на том, что лечение заболевания следует начинать до достижения возраста 10 дней, что для многих стран потребует изменения сроков проведения неонатальных скринингов и ассоциированных с ним процедур. В нашей стране сроки проведения неонатального скрининга соответствуют рекомендациям Европейского руководства по ФКУ (4 сутки жизни у доношенных новорождённых и 7 сутки жизни у недоношенных) [16] .
Тактика контроля фенилкетонурии определяется, в первую очередь, уровнем фенилаланина в крови пациента. Среди специалистов существует единодушное мнение, что уровень фенилаланина более 600 мкмоль/л является прямым показанием к незамедлительному лечению. Кроме нейропедиатра Росы Гассио Субиракс и её соавторов, никто не описывал крупных исследований по изучению необходимости лечения пациентов при уровне фенилаланина менее 360 мкмоль/л [17] . Таким пациентам рекомендовано с особой тщательностью проводить мониторинг данного показателя в течение первого года жизни, так как с возрастом есть риск повышения уровня фенилаланина.
Контроль уровня фенилаланина в крови
Мнение медицинского сообщества относительно начала лечения пациентов с концентрациями фенилаланина 360-600 мкмоль/л достаточно противоречивы. Costello и соавторы [18] проводили исследование, в котором пациенты были разделены на три группы:
- 1 группа — концентрация фенилаланина < 400 мкмоль/л;
- 2 группа — концентрация фенилаланина 400-500 мкмоль/л;
- 3 группа – концентрация фенилаланина > 500 мкмоль/л.
Исследователи обнаружили тенденцию к снижению IQ у лиц с более высоким уровнем фенилаланина и рекомендовали лечение, которое бы обеспечило поддержку уровня фенилаланина в крови < 400 мкмоль/л в течение всего периода детства.
Согласно Российским Федеральным клиническим рекомендациям, при концентрациях фенилаланина более 360 мкмоль/л пациенту назначается лечение.
Диета при фенилкетонурии
На сегодняшний день основным доказанным методом лечения фенилкетонурии считается диетотерапия, которая основывается на трёх подходах:
- Ограничение естественного белка с использованием индивидуальной толерантности к фенилаланину.
- Использование добавок L-аминокислот (не содержащих фенилаланин) для перекрытия физиологических потребностей в микроэлементах.
- Употребление пищи с низким содержанием белка для удовлетворения физиологических потребностей пациента в энергии [19] .
Общее потребление белка должно обеспечивать безопасные уровни потребностей данного макронутриента с дополнительной дотацией 40 % L-аминокислот (20 % L-аминокислот необходимы для компенсации потребностей в незаменимой аминокислоте и еще 20 % L-аминокислот используются для контроля фенилаланина в крови).
При введении неадекватных дозировок L-аминокислот они ограничивают синтез белка. Белковый обмен, который в норме складывается из процессов анаболизма (синтеза) и катаболизма (распада) белков, смещается в сторону катаболизма. При данном процессе фенилаланин остается неиспользованным для синтеза белка, и его концентрация в крови будет расти.
При фенилкетонурии необходимо избегать продуктов, богатых белком (мясо, рыба, яйца, обычный хлеб, большинство сыров, орехи и семена), а также продуктов, содержащих аспартам (подсластитель, который используется при изготовлении некоторых газированных изделий и конфет). Пища с низким содержанием белка при диетотерапии данного заболевания должна содержать 50 мг или менее фенилаланина на 100 г сухого продукта. Фрукты и овощи, содержащие менее 75 мг фенилаланина на 100 г пищевого продукта, также могут быть включены в рацион. При употреблении таких овощей, как картофель, брокколи, цветная капуста, брюссельская капуста, нужно учитывать, что даже их небольшое количество в рационе обеспечивает организм 50 мг фенилаланина, поэтому их потребление не должно быть бесконтрольным. Для удобства родителей и ребёнка следует пользоваться «пищевым светофором», где все продукты разделены на группы, разрешённые (ограниченно или неограниченно) и запрещённые к употреблению.
Когда ребёнок маленький, соблюдение диеты не является проблемой для семьи, так как родители контролируют потребление продуктов. В младенческом возрасте предлагается использовать специализированные смеси без добавок фенилаланина, которые дополняются либо грудным молоком, либо стандартной семью для перекрытия суточной потребности в фенилаланине.
Дети более старшего возраста продолжают не только пить специальный безфенилаланиновый продукт, который способен обеспечить потребности в белках и калориях, но и получают дополнительное количество разрешённых продуктов (овощи и фрукты, мёд, животные и растительные масла, зефир, пастила, варенье), необходимых для создания пищевого разнообразия.
По мере взросления ребёнка соблюдение диеты становится всё труднее, так как дети с фенилкетонурией, в отличие от своих сверстников, значительно ограничены в выборе продуктов, что часто приводит к скачкам концентрации фенилаланина у подростков [20] [21] Долгосрочное поддержание диеты необходимо, поскольку пациенты после периода нарушений диеты намного труднее возвращаются к прежнему режиму питания.
Прогноз. Профилактика
Хороший прогноз для интеллекта возможен в том случае, когда пациенты с первого месяца жизни получают диету с пониженным содержанием фенилаланина. Существует пропорциональная связь между гиперфенилаланинемией и IQ у пациентов с фенилкетонурией, где при повышении данной аминокислоты на каждые 100 мкмоль/л происходит снижение IQ на 1,3-4,1 пункта [15] .
Если пациенты с фенилкетонурией получают и соблюдают лечение с самого раннего возраста, то их качество и продолжительность жизни ничем не отличается от их здоровых сверстников.
Пациенты, не получающие или не соблюдающие лечение, часто имеют инвалидность и низкий уровень качества жизни. Кроме того, несоблюдение диеты и отсутствие контроля фенилаланина в организме часто приводит к снижению продуктивности и внимания, нарушению поведения (особенно при уровне аминокислоты свыше 360 мкмоль/л).
Адекватное наблюдение за концентрацией фенилаланина в пределах допустимых показателей достаточно эффективно в профилактике большинства нарушений центральной нервной системы. Большинство людей демонстрируют нормальное общее развитие, легко справляются с образовательными стандартами, ведут самостоятельную жизнь и получают работу, будучи взрослыми.
При соблюдении пациентом режимов диеты и дополнительной дотации минеральных веществ не отмечается увеличения риска таких осложнений, как остеопороз или частые переломы при отсутствии других заболеваний костно-мышечной системы и соединительной ткани.
Профилактика фенилкетонурии
Если у будущих родителей или их близких родственников выявлена фенилкетонурия, то при планировании беременности рекомендуется проконсультироваться с генетиком и пройти обследование. Так можно определить риск рождения ребёнка с фенилкетонурией.
Чтобы предотвратить врождённые нарушения у ребёнка, женщинам с фенилкетонурией во время беременности следует придерживаться диеты с низким содержанием фенилаланина [22] .
Фенилкетонурия у детей: особенности лечения и рекомендации врачей
Фенилкетонурия у детей относится к редким генетическим патологиям, в основе которой лежит ферментная недостаточность, в частности, невозможность печени усваивать фенилаланин. Вещество поступает в организм вместе с пищей, именно поэтому основным направлением терапии является пожизненная коррекция питания. Неспособность утилизировать фенилаланин в организме приводит к его накоплению в составе крови, отравлению нервной системы и гибели важных нейронов головного мозга. Другое название заболевания – это фенилпировиноградная олигофрения, что объясняет развитие характерной симптоматики. При своевременном выявлении и пожизненном соблюдении лечебного питания ребенок вырастает полноценной личностью с устойчивой психосоматической картиной.

Фенилкетонурия у детей — наследственное заболевание, обусловленное мутацией гена в 12 хромосоме
Механизм формирования
Фенилкетонурия у детей (ФЛК) – наследственно обусловленная болезнь, связанная с нарушением синтеза, распада и эвакуации продуктов аминокислотного обмена из организма, а именно — преобразованием фенилаланина до тирозина. Таким образом, происходит накопление тех веществ, которых в организме быть не должно – ортофенилацитата, фенилмолочной и фенилпировиноградной кислот, фенилэтиламина. Высокие концентрации этих веществ, подобно яду влияют на организм, приводя к следующим состояниям:
- нарушению жирового обмена в головном мозге;
- гибели нейромедиаторов, которые передают импульсы между мозговыми клетками;
- общей интоксикации.
Мишенью при фенилкетонурии является нервная система, поэтому интоксикация приводит к выраженному снижению интеллекта и умственной отсталости — олигофрении.
Заболевание возникает, если носителем мутантного гена являются оба родителя. При этом они могут сами не болеть патологическим синдромом. Риск мутации у ребенка в таком случае достигает 25-30%.
Основные виды
Клиницисты выделяют 3 основных типа патологического состояния у детей:
- I тип. Составляет 95% всех случаев детской фенилкетонурии, обусловлен нарушением синтеза фениланин-4-гидроксилазы.
- II тип. Состояние характеризуется невозможностью выработки фермента дигидроптеридинредуктазы. Возникает в 1-3% среди пациентов с ФЛК. Лечение затруднено, а при его отсутствии наблюдается высокая летальность детей уже в 2-3 года.
- III тип. На фоне патологии наблюдается нарушение синтеза еще одного фермента — тетрагидробиоптерина. Редкая разновидность.
Существуют еще несколько типов заболевания, но они составляют всего 0,1-0,5% от общего числа выявленных случаев.
Причины
Фенилкетонурия характеризуется аутосомно-рецессивным путем наследования. При появлении характерных признаков болезни говорят о мутации гена, который напрямую отвечает за выработку фенилаланина и 12 хромосомы. Отсутствие прямых признаков указывает на развитие ФЛК 2 и 3 типов.
Спровоцировать заболевание способны геномные мутации вблизи 12 хромосомы, перенесенный родителями полиомиелит в раннем детстве, браки между близкими родственниками, системный токсический фактор (например, радиация), муковисцидоз в анамнезе. Повреждение гена может носить спонтанный характер.
Симптомы

Питание — важное условие для поддержания адекватного качества жизни
Сразу после рождения ребенка симптоматический комплекс не выражен или не проявляется вовсе. Первые признаки наблюдаются спустя 2-3 недели или месяц после рождения малыша.
В первые несколько недель жизни заболевание не дает о себе знать, а дети не отличаются от своих сверстников. Начальные признаки нарушения возникают позднее. Среди клинических проявлений у новорожденных выделяют судороги, повышенный мышечный тонус, апатичность, обильные срыгивания, рвоту.
Наиболее характерными для фенилкетонурии считаются следующие поздние признаки:
- заметная и нарастающая умственная отсталость (малыш не интересуется окружающим миром, не узнает окружающих, в позднем возрасте отсутствует способность к обучению, а полученные навыки быстро утрачиваются);
- неприятный запах от кожи (у больных фенилкетонурией наблюдается характерный запах плесени от кожных покровов);
- дерматологические заболевания;
- поздний рост молочных зубов;
- заметное отставание в физическом развитии.
Дети с ФЛК имеют типичную внешность: кожа без пигментации, бледная, глаза и волосы светлые из-за нарушения выработки меланина. Нередко наблюдаются бесконтрольные телодвижения, как при ДЦП.
Диагностика
Фенилкетонурия является областью исследования генетиков, психиатров, невропатологов и гепатологов. Окончательный диагноз ставят на основании следующих данных:
- обнаружение в крови фенилаланина в количестве более 900 мгмоль/л;
- положительный результат пробы Феллинга;
- присутствие в крови фенилмолочной, фенилуксусной или фенилпировиноградной кислоты.
Важными критериями диагностики фенилкетонурии у детей являются жалобы родителей на нетипичное поведение, задержку психофизического развития ребенка, прочие частые при ФЛК симптомы.
Высокой достоверностью обладает ранняя диагностика заболевания, еще в период внутриутробного развития плода. Сегодня каждая женщина, наблюдающаяся у гинеколога по поводу беременности, проходит ряд скрининговых обследований, в том числе и на фенилкетонурию.
Лечение
Лечение фенилкетонурии только консервативное. Оно направлено на стабилизацию метаболических процессов, купирование симптомов, предупреждение олигофрении и других психических расстройств личности. Обязательно назначаются витаминные комплексы, минеральные соединения, лечебная гимнастика, массаж, иглорефлексотерапия.
По показаниям обязателен прием ноотропных, противосудорожных или антиконвульсивных препаратов. При необходимости ребенок наблюдается у дефектолога, логопеда, проводится регулярный анализ крови и электроэнцефалограмма.
Важную роль в терапии фенилкетонурии играет лечебное питание. Учитывая, что фенилаланин проникает в организм с пищей, то коррекция состояния предполагает правильную диету. Основу рациона составляют фрукты, овощи, аминокислотные смеси, гидролизаты. Расширение рациона допустимо только после совершеннолетия, когда наблюдается терпимость организма к фенилаланину.
Особенности питания

Признаки заболевания носят неврологический характер, определяют умственно-физические критерии развития ребенка
Вопрос диеты встает остро уже с самого рождения ребенка. С каждым месяцем при отсутствии терапии и корректирующей диеты наблюдается снижение интеллекта на 3-4 балла по шкале IQ. При уровне фенилаланина в крови до 6% от нормы питание не корректируют. При превышении показателей переходят на безбелковые смеси. Прикорм начинают с овощей, фруктов и соков на их основе. Позже вводят фруктовое пюре из груш, яблок, слив, овощное пюре.
Далее в рацион вводят безмолочные каши, низкобелковые смеси, безбелковый хлеб. Лечебная диета носит пожизненный характер, однако небольшие погрешности допустимы по достижении ребенком совершеннолетия.
Диета при фенилкетонурии исключает следующие продукты:
- птицу, мясо, бульоны на их основе;
- молочные продукты и продукцию на основе молока, включая грудное молоко;
- любую рыбу.
По мере взросления количество суточного белка ребенку увеличивают, но постоянно наблюдают за содержанием фенилаланина в крови. Питание строго контролируют, меню составляют в соответствии с данными энцефалографии, анализа крови. Наряду с повышением суточного белка снижают количество жиров и углеводов. При необходимости получения клинических рекомендаций по поводу лечебного рациона можно обратиться к врачу-гастроэнтерологу, диетологу.
При отмене или несоблюдении правил лечебного питания наблюдается возобновление типичной симптоматики даже на фоне медикаментозной терапии, нарастание умственной отсталости. Диета направлена на поддержание лабораторных показателей в норме, чтобы исключить необратимые изменения в головном мозге.
Осложнения и прогноз
При несоблюдении корректного рациона прогноз по заболеванию крайне неблагоприятный. Ребенок становится умственно отсталым, инвалидом уже через несколько лет. При начале терапии спустя пару лет после рождения могут наблюдаться остаточные расстройства психики, связанные с гибелью клеток головного мозга. Родителям важно соблюдать все клинические рекомендации, объяснять подросшему ребенку опасность несоблюдения диеты.
Массовый скрининг на фенилкетонурию помогает родителям и врачам выстроить адекватную, своевременную тактику ведения больного ребенка, обеспечив ему благоприятные прогнозы по развитию в будущем.
Видео


* Представленная информация не может быть использована для самостоятельной постановки диагноза, определения лечения и не заменяет обращение к врачу!
Источник https://medaboutme.ru/zdorove/spravochnik/bolezni/fenilketonuriya/
Источник https://probolezny.ru/fenilketonuriya/
Источник https://www.baby.ru/wiki/fenilketonuria-u-detej-harakteristika-simptomy-taktika-lecenia-dieta-i-rekomendacii-vracej/




