Фенилкетонурия ( Болезнь Феллинга , Фенилпировиноградная олигофрения )
Фенилкетонурия – это наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
МКБ-10
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных.
В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты. Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов — фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко — врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет. При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным. Основные и дополнительные методы диагностики:
- Скрининг-тест. Проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
- Биохимические исследования. Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др.
- Неврологическое обследование. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
- Пренатальная диагностика. Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза). В остальных случаях окончательный диагноз выставляется по результатам ДНК-диагностики после рождения.
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси — Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет — Максамум-ХР и др. Основу диеты составляют низкобелковые продукты — фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям — ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия. Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Прогноз и профилактика
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Фенилкетонурия

Фенилкетонурия относится к наследственным заболеваниям из группы ферментопатий. Она связана с нарушением обмена ароматических аминокислот, а конкретно — фенилаланина. Если человек с этим заболеванием не соблюдает низкобелковую диету, то в его организме происходит накопление фенилаланина и продуктов метаболизма, обладающих токсическим действием на ЦНС, которое в свою очередь вызывает поражения нервных структур и приводит к нарушениям умственного развития. Возникает фенилпировиноградная олигофрения по аутосомно-рецессивному типу наследования, однако на сегодняшний день это одно из немногих наследственных заболеваний, которое поддается успешному лечению.
Распространённость не зависит от гендерного различия, но отличается у разных групп населения. Однако, риску летального исхода более подвержены мальчики возрастом до 1 года.
В европеоидной расе и среди жителей Америки фенилкетонурия встречается у 1 человека на 10-15 тыс. Наиболее высокая частота случаев зарегистрирована у граждан Турции: 1 из 2,60 тыс. Финляндия и Япония отличается наиболее низкой частотой встречаемости данного недуга не более 1 человека на 120 тыс. рождений. Сенсацией были данные исследований 1987 г, когда среди представителей цыганских популяций в Словакии был обнаружен сверхвысокий уровень фенилкетонурии, вызванный предположительно инбридингом: болезнь выявляли у каждого 4-ого ребенка.
Историческая справка
В 1934 г норвежским врачом Иваром Асбьёрном Феллингом была выявлена гиперфенилаланинемия. Она оказалась ассоциирована с задержкой умственного развития. У жителей Норвегии заболевание называлось в честь его открывателя — болезнь Фёллинга.
Методика успешного лечения фенилкетонурии впервые была разработана в стенах Бирмингемского детского госпиталя (Англия). Ее разработал Хорст Биккель со своей командой медиков в начале пятидесятых годов двадцатого века. Однако, больший успех терапия имела, когда удалось широко применять раннюю диагностику недуга, вызванного повышенным содержанием фенилаланина в кровяном русле новорождённых по методу Гатри, разработанному и внедренному в 1958-1961 годах.
Фенилкетонурия, что это за заболевание?
Название происходит от двух названий химических соединений – фенилаланина и кетонов, а также греческого слова uron означающего – моча. Это заболевание вызывает расстройства движений и тонуса мышц, а также отставание физического развития и прогрессирующее слабоумие, поэтому еще называется фенилпировиноградной олигофренией.
На начальных этапах фенилкетонурия никак себя не проявляет и протекает бессимптомно. Тревожными знаками становится манифестация симптомов в виде сонливости и плохо аппетита у детей 6-12 месяцев. Такие дети отличаются от родственников и ровесников слишком светлой кожей, блондинистыми волосами и голубым цветом глаз. Также характерным симптомом становится развитие сыпи, напоминающей дерматит или экзему. Отсутствие лечения приводит к заметной, значительно выраженной задержке умственного развития, поэтому очень важно выявить болезнь на первых годах жизни, пока не успели произойти необратимые патологические изменения.
Патогенез
Как незаменимая аминокислота – фенилаланин является протеиногенной и играет важную роль в построении белковых молекул всех живых организмов, обеспечивая стабилизацию белковых структур и фолдинг (нативную укладку), являясь составным звеном функциональных центров. Необходим для синтеза гормонов щитовидной железы (в большей мере — тироксина), а также адреналина и меланина.
Метаболический блок превращения фенилаланина в тирозин приводит к его накоплению и активации побочных путей его обмена (например, печенью), в результате чего в тканях организма аккумулируются его токсичные производные — фенилпировиноградная, фениломолочная и другие кетоновые кислоты, в норме которых практически не должно образовываться. Помимо этого, происходит синтез ортофенилацетата и фенилэтиламина – соединений также почти полностью отсутствующих при нормальном метаболизме, причем производные последнего являются психоделиками и психостимуляторами. Наличие их в избытке инициирует нарушение обмена жиров в структурах головного мозга. Считается, что именно этот фактор вызывает прогрессирующее снижение интеллекта и может привести в самой глубокой степени умственной отсталости — идиотии. На сегодняшний день остаются до конца невыясненными механизмы развития нарушений работы мозга, вызванные фенилкетонурией. Предположительно, этому способствует дефицит нейромедиаторов (серотонина, дофамина) в синапсах мозга на фоне прямого токсического действия фенилаланина и относительного снижения количества тирозина и прочих «крупных» аминокислот, способных конкурировать с фенилаланином в процессах переноса через гематоэнцефалический барьер.
Гиперфенилаланинемия приводит к нарушениям обмена липо- и гликопротеидов, аминокислотного равновесия как жидкостных сред, так и клеточных структур. В основе клинических проявлений также лежат нарушения метаболизма гормонов, обмена моноаминовых нейромедиаторов таких как катехоламины и серотонин.
Разлад функции печени выражен в виде диспротеинемии, генерализованной гипераминоацидемии, метаболического ацидоза, нарушения белковосинтезирующей и окислительной деятельности клеточных органелл.
Классификация
Различают типичную (классическую) и атипичные формы (варинатные, коферментзависимые, злокачественные — на них приходится примерно 10% случаев) фенилкетонурии в связи с генетической и клинической неоднородностью аномалий обмена фенилаланина, а также кофактора — биоптерина.
Причиной второй диеторезистентной атипичной формы фенилкетонурии является дефицит дигидроптеридинредуктазы в результате генного дефекта локализующегося на участке 4р 15.3 хромосомы. Открыл ее Смит в 1974 году. Энзимный дефект нарушает процесс восстановление активной формы тетрагидробиоптерина, принимающего участие в виде кофактора при гидроксилировании фенилаланина в тирозин, а также предшественников нейромедиаторов серотонинового и катехоламинового ряда, таких как L-ДОФА и 5-окситриптофан.
Фенилкетонурию 3 диеторезистентную описал Кауфман в 1978 г. (согласно источнику Википедия). Она отличается недостаточностью 6-пирувоил тетрагидроптерин синтетазы, принимающей участие в синтезе тетрагидробиоптерина.
Атипичные формы отличаются прогрессированием проявлений заболевания.
Другие формы патологии связаны с нарушением иных альтернативных путей метаболизма фенилаланина с формированием метилминдальной ацидурии и парагидроскифенилуксусной ацидурии.
Причины возникновения фенилкетонурии
Благодаря накоплению опыта в области диагностики и лечения фенилкетонурии с течением времени стало понятно, что причины возникновения этого наследственного недуга кроются в мутациях гена, кодирующего фенилаланин-4-гидроксилазу — 12q23.2. Тип наследования аутосомно-рецессивный.
Чаще всего болезнь вызвана резко сниженной или полностью отсутствующей активностью такого фермента печени как фенилаланин-4-гидроксилаза, которая в норме необходима для катализации превращения (гидроксилирования) фенилаланина в аминокислоту — тирозин.
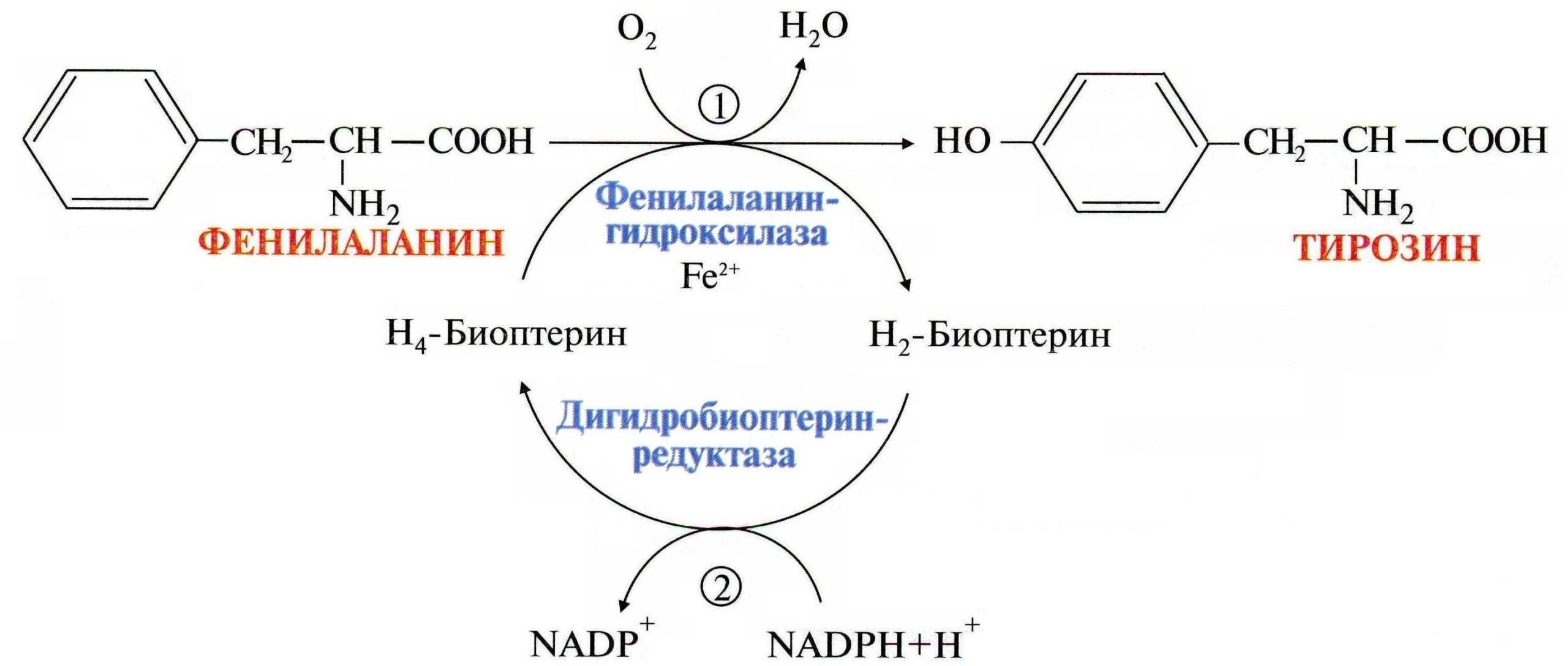
Обмен фенилаланина и тирозина
Однако, примерно в 10% случаев гиперфенилаланинемии воздействую атипичные факторы, связанные с мутациями в иных генах, кодирующих ферменты, обеспечивающие синтез кофактора фенилаланингидроксилазы, известного как тетрагидробиоптерин (BH4).
Симптомы фенилкетонурии
Фенилкетонурия отличается достаточно ярко выраженной клинической картиной, включающей такие симптомы как:
- отставание физического развития с 6 месячного возраста;
- микроцефалия;
- вегетативные дисфункции;
- повышенная возбудимость и двигательная гиперактивность; или дерматит, возможно с папулезными кожными высыпаниями (как на фото ребенка с фенилукетонурией);
- мышечная гипертензия;
- атаксия;
- гиперкинезы;
- неустойчивость походки;
- судорожные припадки;
- нередко обнаруживаются пороки сердца;
- чувствительность к травматизации и осветление кожи, волос и радужки глаз (депигментация), в особенности при несоблюдении необходимой диеты, вызывающей недостаточность меланина в организме, являющегося производным тирозина.
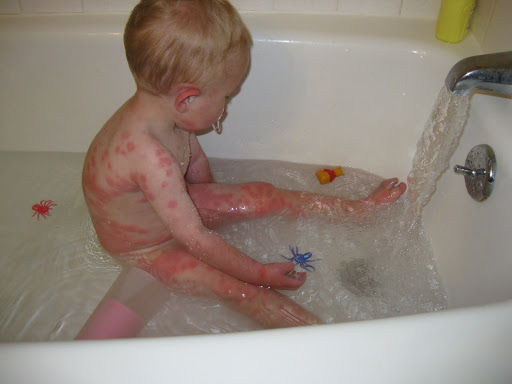
Экзема при фенилкетонурии
Психические нарушения
Фенилкетонурия вызывает значительные патологические изменения обменных процессов в головном мозге, что влечет за собой следующие нарушения:
- глубокая степень умственной отсталости, вплоть до идиотии или имбецильности;
- трудности в обучении;
- возникновение явлений эхопраксии — повторение движений за окружающими;
- эхолалии (повторение речи);
- вялое поведение может сменяться редкими вспышками злости и раздражительности.
Как можно увидеть на фото больных фенилкетонурией телосложение их обычно диспластическое, размеры черепа уменьшены, отмечается гипогенитализм и нанизм.
Анализы и диагностика
В основе диагностики фенилкетонурии лежат лабораторные анализы крови и мочи, изучают биоптаты печени, а также проводят генетические исследования:
- полуколичественный тест или количественное определение фениаланина в кровяном русле, в норме составляет 0,01-0,02 г/л, но при фенилкетонурии присутствует во всех жидких средах организма, а в сыворотке крови повышается до 0,15-0,2 г/л;
- на ранних этапах жизни (у детей 10-12 дней) могут быть выявлены в моче продукты распада фенилаланина – фенилкетоны, в особенности при нелеченой клинической картине, при этом используется проба Феллинга с 5-10% раствором FeCI3 или индикаторные бумажки «Фенистикс» и «Биофан», меняющие при положительном результате цвет на сине-зеленый;
- в печеночном биоптате исследуют уровень активности фенилаланингидроксилазы;
- проводят поиск мутаций в хромосомах гена фенилаланингидроксилазы, тогда как для 2 и 3 типа заболевания, связанного с мутациями в гене, отвечающем за биосинтез кофактора необходимо выполнение дополнительных диагностических исследований – выявления в моче низкого количества продуктов обмена нейротрансмиттеров, таких как гомованилиновая, ванилилминальная и оксииндолилуксусная кислоты.
На 3-4 сутки жизни новорожденного наиважнейшим в обнаружении обменных врождённых заболеваний оказывается неонатальный скрининг посредством анализов крови. Этот этап делает возможным обнаружить фенилкетонурию и как можно раньше начать лечение, чтобы предотвратить необратимые последствия. В РФ таким детям устанавливается категория инвалидности до 18 лет.
Лечение фенилкетонурии
Наиболее благоприятным оказывается прогноз при ранней диагностике заболевания. Если же недуг выявляется поздно, то практически невозможно справиться с уже резвившимися необратимыми изменениями тканей мозга.
В основе лечения фенилкетонурии – строгая диета, ограничивающая животные и растительные белки, которая должна длиться минимум до полового созревания, а может и пожизненно. Постоянно нужно быть в контакте с лечащим врачом, который будет мониторить состояния и психологически поддерживать пациента. При отмене диеты регулярно проводят психологические тесты, электроэнцефалограмму и определяют уровень фенилаланина в кровяном русле. Процесс ослабления диеты начинают примерно в 8-10 лет, когда заканчиваются процессы миелинизации мозга.
В случаях некоторых (мягких) форм заболевания возможно лечение кофактором (тетрагидробиоптерином) при поражённом ферменте — фенилаланингидроксилазе. Перспективными считаются разработки новых подходов лечения фенилкетонурии — использования заместительной терапии с применением фенилаланинлиазы (PAL, пегвалиазы) — растительного фермента, завершающего метаболизм фенилаланина безвредными метаболитами, а также генотерапия, в основе которой введение в организм вирусного вектора с геном фенилаланингидроксилазой. Такие методы не смогли пока выйти из стен лабораторных исследований.
При атипичных формах, не поддающихся диетотерапии, лечение сводится к введению препаратов тетрагидробиоптерина либо его синтетических аналогов, к примеру, сапроптерина.
Наиболее перспективной методикой лечения данного тяжелого заболевания является генотерапия как классический образец успешной борьбы и организационной помощи при наследственных патологиях.
Фенилкетонурия
Фенилкетонурия (ФКУ) – это врожденная аутосомно-рецессивное заболевание из группы ферментопатий (заболеваний, при которых нарушается работа ферментов), связанное с нарушением обмена аминокислоты фенилаланина. Фенилаланин является незаменимой аминокислотой, поступающей в организм человека с белковой пищей. В организме человека фенилаланин используется для синтеза белков, а неиспользованный запас превращается в аминокислоту тирозин. При фенилкетонурии обмен аминокислоты нарушается, и ее уровень начинает повышаться.
Заболевание встречается у 1 ребенка на 10 000 новорожденных с одинаковой частотой среди мальчиков и девочек.

Суперфуды в косметике: сочные коктейли для здоровья кожи и волос
Симптомы
Клиническая картина фенилкетонурии развивается в первом полугодии жизни и с ростом малыша прогрессирует. На первых месяцах жизни ребенок становится вялым, либо появляются раздражительность, плаксивость, беспокойство, отсутствие интереса к окружающему миру, игрушкам. Он отстает в психическом, умственном развитии, замедляется рост черепа. Для таких детей характерен специфический затхлый «мышиный» запах тела.
Основные предъявляемые жалобы (со слов родителей): при отсутствии лечения на первом году жизни, обычно в возрасте 2−6 месяцев, родителей беспокоят срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония – слабость мышц), признаки атопического дерматита, задержка психомоторного развития, иногда судороги.
Ребенок поздно учится сидеть, ползать, ходить, зубы прорезываются позже, чем у сверстников. Формируется специфическая поза и осанка при сидении. Нередко могут присоединиться симптомы поражения центральной нервной системы: навязчивые состояния, судороги, тремор (дрожание конечностей) с дальнейшим формированием эпилепсии. Как правило, дети голубоглазые, светловолосые, кожа светлая (содержит мало пигмента меланина), сухая с шелушением. У темнокожих и темноволосых детей, заболевание встречается очень редко.
Формы
Классификация классической фенилкетонурии по форме заболевания исходя из содержания фенилаланин в крови: легкая гиперфенилаланинемия (не ФКУ) — содержание фенилаланина 120−600 мкмоль/л; умеренная (мягкая, средняя) ФКУ — содержание фенилаланина 600−1200 мкмоль/л; классическая (тяжелая) ФКУ, когда уровень фенилаланин превышает 1200 мкмоль/л.
Причины
Основной причиной развития данной патологии является отсутствие или очень низкая выработка печеночного фермента фенилаланингидроксилазы, который превращает фермент фенилаланин в тирозин. В результате отсутствия данного фермента в организме начинает накапливаться фенилаланин и его производные, которые оказывают токсическое воздействие на организм ребенка, в частности, на центральную нервную систему.
Методы диагностики
Диагностика фенилкетонурии осуществляется врачом-педиатром на основании сбора жалоб, данных анамнеза, клинического осмотра, лабораторных (включая генетические) и дополнительных инструментальных методов обследования. Диагностика направлена на определение клинической формы заболевания, его причины, тяжести состояния и возникающих осложнений.
Все новорожденные дети уже в роддоме на 3-4 сутки (недоношенным на 7-е сутки) по программе неонатального скрининга обследуются на фенилкетонурию. Содержание фенилаланина выше 2,0 мг/дл классифицируется как гиперфенилаланинемия, которая требует проведения уточняющей диагностики. Определяют концентрацию фенилаланина в сухих пятнах крови новорожденных методом флуориметрии (метод определения концентрации вещества по интенсивности флуоресценции, возникающей при облучении вещества монохроматическим излучением) и методом тандемной масс-спектрометрии (метод исследования вещества, основанный на определении отношения массы к заряду ионов,).
В настоящее время известно более 900 мутаций в гене РАН, которые могут привести к развитию фенилкетонурии: поэтому показано генетическое исследование. В биохимическом анализе крови и мочи может определяться снижение содержания гомованилиновой кислоты и 5-оксииндолуксусной кислоты, содержание кетоновых тел повышено.
Для выявления изменений в различных органах и наличии осложнений назначают проведение инструментальных методов исследования – электроэнцефалография, МРТ – магнитно-резонансная томография с целью выявления очагов поражения коры мозга и других изменений у пациентов старше 12 лет; УЗИ брюшной полости и почек для диагностики дискинезии (нарушения моторики) желчных путей, диффузных изменений печени и поджелудочной железы, мочекаменной болезни; фиброгастродуоденоскопия (проводится по показаниям) – для диагностики поражения слизистой оболочки желудка.
Дифференциальная диагностика фенилкетонурии проводится с транзиторной гиперфенилаланинемией недоношенных, тирозинемией (нарушения метаболизма с повреждением печени, почек, периферических нервов), галактоземией (наследственное заболевание, в основе которого лежит нарушение обмена веществ на пути преобразования галактозы в глюкозу), другими заболеваниями, связанными с нарушением функции печени.
Основные используемые лабораторные исследования:
- Определение концентраций фенилаланина в сухих пятнах крови методом флуориметрии (в первые дни жизни).
- Фенилаланин в крови (тандемная масс-спектрометрия) – увеличение;
- Проба Фелинга (определение фенилпировиноградной кислоты в моче) – положительна.
- Генетическое исследование мутации гена PAH: мутация R408W, мутации IVS12nt1, R261Q, R252W, R158Q, P281L, IVS10nt546, I65T (предрасположенность к развитию фенилкетонурии).
Дополнительные используемые лабораторные исследования:
- Птерины в моче: снижение образования белка биоптерина (кофермент, участвующий в ряде важных биохимических реакций) при мутации гена PTS; при мутации в гене DHPR – образование биоптерина повышено.
- Кетоновые кислоты в моче повышены.
- Гомованилиновая кислота, 5-оксииндолуксусной кислота (в крови) повышены.
- Генетическое исследование на гиперфенилаланемию – гены: PAH, GCH1, DHPR, PCBD1, PTS, QDPR.
Основные используемые инструментальные исследования:
- Электроэнцефалография;
- МРТ головного мозга (очаги повреждения коры головного мозга);
- УЗИ органов брюшной полости;
- Эзофагогастроскопия (диагностика поражения слизистой желудка).
Лечение
Лечение фенилкетонурии заключается в — снижении фенилаланина в крови, повышении толерантности (переносимость) фенилаланина, получаемого с натуральной пищей. И таким образом избежать тяжелой неврологической симптоматики и улучшить качество жизни.
Диетотерапия с исключением продуктов содержащих фенилаланин (рыбные, молочные, мясные продукты). Категорически запрещается использование газированных напитков, содержащих фенилаланин. Для вскармливания грудных детей разработаны специальные смеси с низким содержанием фенилаланина. Своевременное выявление патологии и соблюдение диеты позволяет избежать развитие необратимых процессов со стороны центральной нервной системы.
Все дети с фенилкетонурией подлежат наблюдению участковым педиатром и психоневрологом с проведением контроля уровня в крови фенилаланина.
Осложнения
Возможны развитие микроцефалия (маленький размер черепа), задержка умственного развития или умственная отсталость, олигофрения.
Профилактика
Профилактика фенилкетонурии включает несколько мер:
- Медико-генетическое консультирование пар с рекомендацией обследования на гетерозиготное носительство частых мутаций в гене РАН;
- Неонатальный скрининг: лабораторное обследование для своевременного выявления и начала лечения больных детей с целью предотвращения их инвалидизации;
- Пренатальная диагностика фенилкетонурии в семье, где уже есть ребенок с фенилкетонурией (выявления фенилкетонурии на стадии внутриутробного развития путем определения активности фенилаланингидроксилазы в культуре амниоцитов (клеток плодного пузыря);
- Профилактика рождения детей с синдромом «материнской фенилкетонурии» от женщин, больных ФКУ, путем организации психологической помощи девочкам-подросткам по вопросам необходимости соблюдения строгой гипофенилаланиновой диеты в пубертатный период, а также консультативной помощи по вопросам планирования семьи и беременности.
Какие вопросы следует задать врачу
Влияет ли фенилкетонурия на развитие детей?
Как можно выявить фенилкетонурию?
Существует ли лечение фенилкетонурии?
Советы пациенту
Поскольку фенилаланин составляет 4-6% всех пищевых белков, продукты с высоким содержанием белка, такие как мясо, рыба, яйца, сыр, орехи, соя, как правило, не разрешается употреблять пациентам с высокой степенью тяжести фенилкетонурии. Потребность в белках и азоте обеспечивается при помощи белковых заменителей, в которых не содержится фенилаланин. Необходимо обеспечение организма всеми витаминами, минералами, незаменимыми жирными кислотами и ненасыщенными жирными кислотами с длинной цепью для поддержания потребности в питательных веществах.
Источник https://www.krasotaimedicina.ru/diseases/children/phenylketonuria
Источник https://medside.ru/fenilketonuriya
Источник https://medaboutme.ru/zdorove/spravochnik/bolezni/fenilketonuriya/

